2.1 细胞长、短轴末端的模量分布特征,肌动蛋白、肌球蛋白Ⅱ异常引起细胞表面弹性模量的非对称升降 采用原子力显微镜测量平面基底上细胞长轴末端与短轴末端的表面弹性模量,测量时间跨度为60 min。将测量结果中各序列最低点的模量值设置为1,所得某一个细胞的极区弹性模量随时间的变化结果如图2所示,可以看出,正常贴壁生长的细胞长、短轴末端的表面弹性模量均在一定范围内波动,且波动的范围较大。如图2A所示,细胞表面弹性模量波动比例大致分布在最低值的两三倍(以最低值为1),但是均属于随机涨落,没有明显的数值变化方向。这与VARGA等[12]的报道一致。
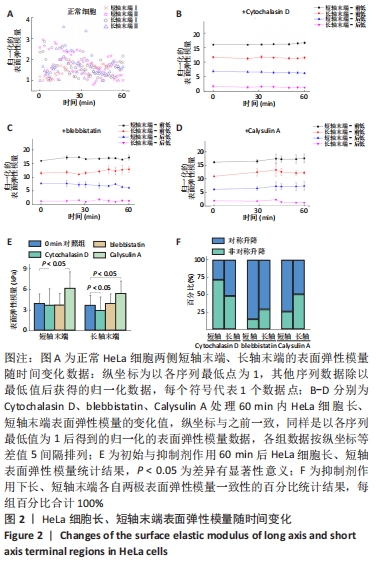
采用肌动蛋白纤维聚合抑制剂Cytochalasin D(2.0 μmol/L)、肌球蛋白Ⅱ的ATP酶活性抑制剂blebbistatin(80 μmol/L)以及肌球蛋白Ⅱ收缩活性增强剂Calysulin A (20 nmol/L)分别作用于HeLa细胞。Calysulin A是1型和2A型蛋白磷酸酶抑制剂,能抑制肌球蛋白Ⅱ的去磷酸化从而提高其活性[17-18],测量开始作用后60 min的极区表面弹性模量变化。以各组模量的最低值为基准1。由于测量结果中的模量最低点出现位置不统一,将30 min前出现最低点的情况归为一组,命名为“前低”,将30 min及之后出现低点的情况归为一组,命名为“后低”。为便于对比并显示统计方差结果,将各点的结果按等差值分布于纵坐标排列,所得结果如图2B-D所示。
与正常组相比具有2个重要的特征差异:①抑制剂处理组使细胞的长轴与短轴末端模量变化趋势与正常组不同;②各抑制剂处理组均出现对侧末端模量最低点分布相反以及模量值升高/降低方向相反的情况,作者认为这种长、短轴末端的模量变化差异可以理解为细胞在骨架功能受限状态下的普遍调节机制。
此外,抑制剂处理组结果显示,细胞在肌动蛋白纤维受抑制情况下,细胞极区末端模量会显著下降,但是仍有部分细胞的单侧短轴末端的模量随时间上升。与之相反,肌球蛋白 Ⅱ收缩活性增强组表现为极区末端模量上升(其中在统计中仅有一侧长轴末端出现下降)。肌球蛋白Ⅱ收缩活性抑制组极区末端模量结果与肌动蛋白更为类似,只是有一侧长轴的统计数据没有明显趋势。
抑制剂处理前后细胞长、短轴末端表面弹性模量值,如图2E所示,Cytochalasin D抑制肌动蛋白纤维聚合使细胞表面弹性模量下降,长轴末端的表面弹性模量较正常细胞下降显著(P < 0.05);blebbistatin抑制肌球蛋白Ⅱ的ATP酶活性分别使细胞长轴末端模量上升和细胞短轴末端模量下降,但作用效果不显著(P > 0.05);Calysulin A增强肌球蛋白Ⅱ收缩活性使长、短轴末端模量显著上升(P < 0.05)。与图2B-D的结果对比可知,Calysulin A增强肌球蛋白Ⅱ收缩的作用效果是显著增强表面弹性模量,而blebbistatin抑制肌球蛋白Ⅱ收缩功能对表面弹性模量整体影响较小;Cytochalasin D抑制肌动蛋白纤维使模量整体降低,但短轴末端模量在这一过程中却明显升高,只抑制肌动蛋白聚合并不能全面降低细胞的表面弹性模量。
细胞对应两侧极区模量变化的一致性百分比,将对侧两端模量最终同向升高/降低定义为对称升降,对侧两端模量最终出现反向升高/降低定义非对称升降,如图2F所示,blebbistatin抑制肌球蛋白Ⅱ收缩活性组对称升降最好,而Cytochalasin D抑制肌动蛋白纤维组的对称升降最差,表明当肌动蛋白纤维聚合受限制,细胞通常会出现在对应两侧力学性能非对称调节,这与图2B-D的分析一致。
从模量的变化值来看,作者认为细胞的弹性模量降低会导致细胞更倾向于通过调节自身结构和蛋白的方式增强极区的模量以稳定自身结构和功能,导致更多的细胞出现明显的力学性能非对称升降。而细胞的弹性模量变化不大或升高时,多数细胞则不需要以力学性能非对称升降做为调节方式。
2.2 肌动蛋白与肌球蛋白Ⅱ在细胞内的分布与活化特征 为进一步阐明此调节对肌动蛋白、肌球蛋白Ⅱ骨架的依赖方式,对这两种蛋白以及调节肌球蛋白Ⅱ的Rho A进行分析。RhoA活化是通过ROCK促进肌球蛋白轻链磷酸化,从而促进细胞的收缩[19-20]。由于实验关注的细胞长、短轴末端位于细胞边缘,所以选取细胞中间层的光学切面进行蛋白分布与活化检测,以更好地展示细胞边缘特征,如图3,4所示。
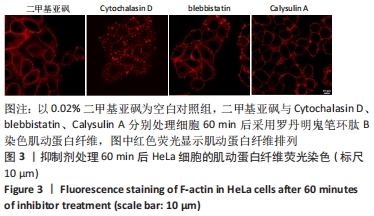
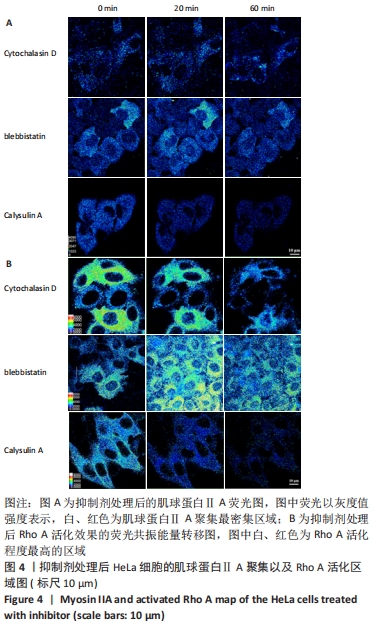
图3为肌动蛋白纤维在抑制剂处理60 min后的结果,Cytochalasin D的作用效果导致细胞肌动蛋白呈区域状分布,没有明显纤维状结构,其他各组在细胞边缘均有纤维状排列的肌动蛋白丝,但Calysulin A处理组不同于空白对照组和blebbistatin处理组,表现为肌动蛋白纤维全部集中在细胞外轮廓。结合之前的数据,作者认为肌动蛋白纤维异常集中于边缘轮廓导致细胞表面弹性模量显著升高。
图4A展示肌球蛋白ⅡA分布密度,Cytochalasin D、blebbistatin以及Calysulin A处理组的聚集区域随时间产生的改变均不显著,但是Cytochalasin D处理组60 min时在细胞边缘及长短轴末端仍出现聚集区域。Calysulin A的处理结果认为增强细胞表面弹性模量的是肌球蛋白Ⅱ收缩活性,而非肌球蛋白Ⅱ的聚集,因为Calysulin A处理组没有发生肌球蛋白Ⅱ聚集的改变。从结果反而推测肌球蛋白ⅡA的聚集有可能会降低表面弹性模量,这需要今后实验的辨别。
进一步测量Rho A的活化分布,分析细胞对肌球蛋白ⅡA的调节可能产生的影响,如图4B所示,Cytochalasin D处理组的Rho A活化减弱,最终Rho A仅在细胞边缘区域有低水平的活化;blebbistatin处理组出现Rho A活化先显著增强(背景位置细胞的显著活化),后随时间延长而再次减弱,但活化区域没有改变;Calysulin A处理组的Rho A活化整体显著降低,但没有区域性改变。由此作者认为,Rho A活化增强肌球蛋白Ⅱ收缩的调节功能是细胞维持自身模量的一种重要手段。当模量升高时(Calysulin A处理组),细胞会显著降低Rho A活化以降低肌球蛋白Ⅱ收缩活性抵抗过高的模量出现,这是一种负反馈调节机制;对于blebbistatin处理组,Rho A主动抵抗肌球蛋白Ⅱ的收缩活性降低而表现出高度活化,可能正是这种负反馈机制导致细胞在blebbistatin作用下没有出现模量的显著下降。当模量降低时(Cytochalasin D处理组),Rho A并未表现出剧烈的活化,只是维持了细胞边缘区域的少量活化,表明在抵抗低模量时,细胞不是通过Rho A调节肌球蛋白Ⅱ收缩活性。
总之,荧光分析实验结果显示,肌动蛋白向细胞边缘明显聚集,会显著增强细胞表面弹性模量,肌球蛋白Ⅱ影响表面弹性模量的方式不是通过区域性聚集,而是通过肌球蛋白Ⅱ的收缩活性来调节。