中国组织工程研究 ›› 2025, Vol. 29 ›› Issue (7): 1414-1421.doi: 10.12307/2025.025
• 细胞相关实验/试验研究Cell related experimental/trial studies • 上一篇 下一篇
过表达溶质载体家族1成员5和敲低慢病毒载体构建及稳定转染RAW264.7细胞株
郭大鑫1,范苏苏2,朱振东2,侯建红1,张 旋2
- 1云南省第三人民医院/大理大学第二附属医院,云南省昆明市 650011;2昆明医科大学药学院暨云南省天然药物药理重点实验室/现代生物医药产业学院,云南省昆明市 650500
Construction of lentiviral vectors for solute carrier family 1 member 5 overexpression and knockdown and stably transfected RAW264.7 cell line
Guo Daxin1, Fan Susu2, Zhu Zhendong2, Hou Jianhong1, Zhang Xuan2
- 1Third People’s Hospital of Yunnan Province /Second Affiliated Hospital of Dali University, Kunming 650011, Yunnan Province, China; 2School of Pharmaceutical Sciences & Yunnan Key Laboratory of Pharmacology for Natural Products/College of Modern Biomedical Industry, Kunming Medical University, Kunming 650500, Yunnan Province, China
摘要:

文题释义:
慢病毒载体:慢病毒是一类基因工程工具,主要用于将目的基因稳定地整合到宿主细胞的基因组中。与常规的腺病毒或其他病毒相比,慢病毒具有高效、稳定的基因转移能力。在有关于溶质载体家族1成员5(SLC1A5)的研究中,使用慢病毒的目的是为了确保目的基因在RAW264.7细胞中稳定表达,为后续的机制探索和治疗策略的验证提供坚实的实验基础。RAW264.7细胞株:RAW264.7细胞是来源于小鼠巨噬细胞的一种常用细胞株。由于易于培养、稳定性好以及具有典型的巨噬细胞特性,被广泛应用于免疫学、肿瘤生物学等领域的研究中。通过在RAW264.7细胞中稳定表达或敲低SLC1A5,研究者可以更深入地探究其对巨噬细胞功能、代谢和免疫应答的影响,为SLC1A5在免疫调节和疾病发生发展中的作用提供有力的实验工具。
背景:溶质载体家族1成员5(solute carrier family 1 member 5,SLC1A5)在多种疾病中发挥了潜在作用,但确切作用机制尚不清楚。构建稳定的SLC1A5过表达和敲低细胞模型可为深入研究SLC1A5在疾病中的确切作用机制以及发现潜在治疗靶点提供有力的实验工具。
目的:构建小鼠SLC1A5过表达和敲低的慢病毒载体,以建立稳定转染的RAW264.7细胞株,为深入探讨SLC1A5在炎症中的作用提供实验基础。
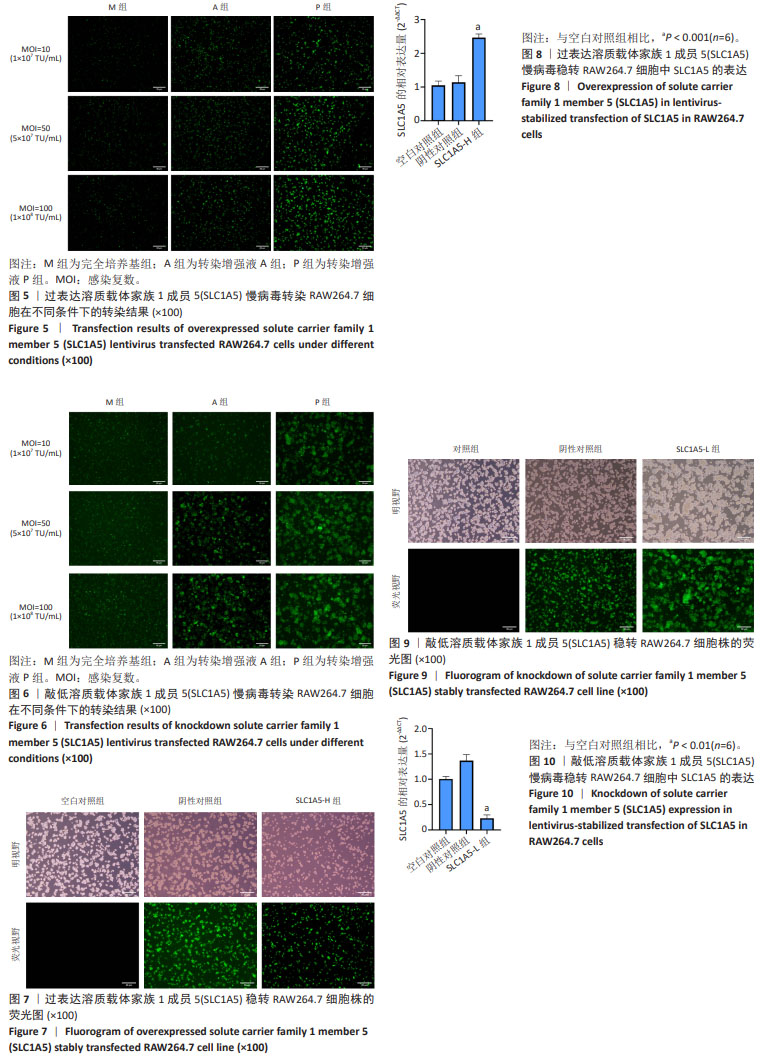
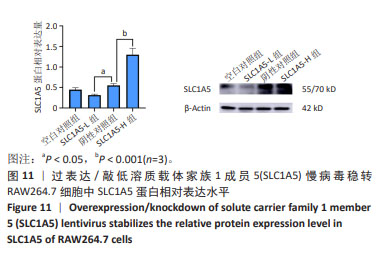
方法:根据SLC1A5基因序列设计合成引物并使用聚合酶链反应扩增该基因片段。将目的基因定向接入经AgeI/NheI酶切的载体质粒GV492中构建重组慢病毒质粒,对阳性克隆进一步筛选后测序比对结果;pHelper1.0质粒载体、pHelper2.0质粒载体、目的质粒载体与293T细胞共同培养并转染,获得慢病毒原液进行包装和滴度测定;在此基础上,通过体外培养RAW264.7细胞,确定嘌呤霉素工作质量浓度;不同滴度的慢病毒分别与RAW264.7细胞共同培养,根据荧光强度确定转染效率;用嘌呤霉素挑选出稳定转染细胞,实时荧光定量聚合酶链反应和蛋白免疫印迹方法检测稳定转染细胞株的SLC1A5基因和蛋白表达水平。
结果与结论:①测序序列与目的序列一致提示重组慢病毒载体构建成功;②过表达SLC1A5慢病毒的滴度为1×109 TU/mL,敲低SLC1A5慢病毒的滴度为3×109 TU/mL;③确定RAW264.7细胞嘌呤霉素工作质量浓度为3 μg/mL;④过表达/敲低SLC1A5慢病毒转染RAW264.7细胞的最佳条件皆为HiTransG P转染增强液且感染复数值等于50;⑤过表达SLC1A5稳转细胞株中SLC1A5基因和蛋白的表达量明显上调,而敲低SLC1A5稳转细胞株中SLC1A5基因和蛋白的表达量显著下调。结果表明,成功构建了小鼠SLC1A5过表达和敲低的慢病毒载体并获得稳定转染的RAW264.7细胞株。
中国组织工程研究杂志出版内容重点:干细胞;骨髓干细胞;造血干细胞;脂肪干细胞;肿瘤干细胞;胚胎干细胞;脐带脐血干细胞;干细胞诱导;干细胞分化;组织工程
中图分类号: