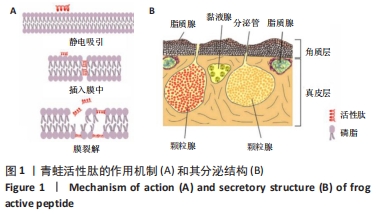
1.1 设计 通过分子生物学方法得到一种青蛙皮肤活性肽序列并合成评价其抗菌抑癌活性。
1.2 时间及地点 实验于2021年6月至2022年6月在英国贝尔法斯特女王大学药学院天然药物研究组完成。
1.3 材料
1.3.1 菌种和细胞 金黄色葡萄球菌S. aures (ATCC CRM6538),大肠杆菌E. coli (ATCC CRM8739),白色念珠菌C. albicans (ATCC CRM10231),感受态大肠杆菌E. coli (JM109 High-Efficiency Competent Cells,Promega,USA)存于英国贝尔法斯特女王大学药学院天然药物研究组菌种保存室。人非小细胞肺癌细胞NCI-H838存于英国贝尔法斯特女王大学药学院天然药物研究组细胞保存室。
1.3.2 主要试剂 Dynabeads®mRNA DIRECTTM试剂盒(Dynal Biotech Ltd,UK);BD SMARTTM RACE cDNA扩增试剂盒 (BD Biosciences,UK);BD SMARTTM RACEDNA扩增试剂盒 (Clontech,UK);琼脂糖、TBE缓冲液、DNA ladder、Tris/Borate/EDTA缓冲液、SYDR safe DNA gel、锥虫蓝(Invitrogen,UK);E.Z.N.A® Cycle Pure Kit (Omega Bio-Tek,USA);pGEM®-T和pGEM®-T Easy Vector kit (Promega,USA);Big Dye®Terminator v31循环测序试剂盒(Applied Biosystems,USA);DNA ladder、RPMI-1640培养基(Invitrogen,Paisley,UK);MTT (Sigma,UK)。
1.3.3 主要仪器 紫外凝胶成像仪(NVP,Nuffield Road,Cambridge,UK);ABI3100自动测序仪(Applied Biosystems,USA);分布式自动固相合成器(Protein Technologies,Inc,Tucson,AZ,USA);反相高效液相色谱柱Jupiter C5柱(250 mm×4.6 mm,Phenomenex,UK);基质辅助激光解吸飞行时间质谱仪(Voyager DE,PerSeptive Biosystems,Framingham,MAUSA);高速离心机(Eppendorf,Germany);自动固相肽合成仪 (Protein Technologies,Tucson,AZ,USA);Cecil Adept CE4200 高效液相色谱仪(Amersham Biosciences);多功能酶标仪 (BioTek, USA)。
1.4 实验方法
1.4.1 红眼树蛙皮肤分泌物的收集提取 获得红眼树蛙(商业渠道,英国)。在模拟生活环境(20-25 ℃,12 h/12 h的光/暗循环)中,将青蛙饲养在动物房,每周喂食3次。饲养4个月后,通过电刺激(6 V,140 ms 脉冲间隔,100 Hz)持续20 s,获得蛙皮分泌物。用ddH2O洗涤收集,在液氮中冷冻,冻干。以上所有程序对实验动物无害,这项研究是按照《1986年英国动物(科学程序)法》的指导下进行的,符合动物伦理。
1.4.2 分子克隆
mRNA的分离:将质量为5 mg的冻干皮肤分泌物溶解在1 mL溶解/结合缓冲液中,并涡旋15 min,13 000×g离心10 min,悬浮液澄清后,丢弃上清液。将体积为250 μL的磁珠放入无RNase离心管中,并转移至磁力架。向无RNase离心管中加入等体积的250 μL裂解/结合溶液,以清洗珠子。将离心管从磁力架上移出,然后将溶解的样品溶液加入这些离心管中。将磁珠与1 mL溶解的样品溶液混合,并将管涡旋10 min以完全分散磁珠。将离心管放置在磁力架上2 min,并去除上清液。磁珠/mRNA复合物在室温下用500 μL洗脱液B洗涤2次。在每个洗涤步骤之间用磁力架从悬浮液中分离珠子。将洗脱液18 μL Tris-HCl(pH=7.5)(10 mmol/L)添加到珠子/mRNA管中,并在80 ℃下孵育2 min。将离心管立即放置在磁力架上,将含有mRNA的上清液转移到新的无RNase离心管中。将新的离心管置于冰上,解开poly-A和poly-T的链以分离mRNA。
构建cDNA文库:使用扩增试剂盒通过反转录聚合酶链反应,将mRNA作为模板合成单链cDNA。为了合成3’-RACE-Ready cDNA,可以使用通过氢键与mRNA结合的3’-CDS引物扩增互补链。首先,合成RACE-Ready cDNA。混合3’-RACE Ready cDNA合成反应溶剂体系:5×第一链试剂2 μL,二硫苏糖醇 (100 mmol/L)1 μL,dNTPs(20 mmol/L)1 μL和反转录酶1 μL。将3’-RACE Ready cDNA PCR试剂(mRNA 4 μL,3’-CDS Primer 1 μL)混合在0.2 mL PCR管中,并标记、涡旋在室温下放置。将离心管在70 ℃孵育2 min以将引物与mRNA结合,然后在冰上冷却2 min。14 000×g离心10 min,收集瓶子底部的内容物。将体积为5 μL的Master Mix试剂添加到上述含有变性RNA的离心管中,14 000×g离心10 min,收集管底部的内容物。将离心管在42 ℃下孵育90 min,以开始从3’端到5’端的转录。用50 μL水稀释第一链cDNA合成反应产物。将离心管在72 ℃加热7 min以结束转录过程。
cDNA末端的快速扩增(3’-RACE PCR):制备PCR Master Mix用于PCR扩增反应,溶剂体系如下:H2O 2.6 μL,10×BD PCR溶剂 1.0 μL,dNTP(10 mmol/L) 0.2 μL,NUP引物 (100 mmol/L) 0.5 μL,50×BD聚合酶0.2 μL,上游引物(20 mmol/L) 0.5 μL。上游引物:5’-GCT CTG WMA TGA CTG TGG TT-3’,W=A+T,M=A+C;通用引物序列:5’-AAG CAG TGG TAT CAA CGC AGA GT-3’。
在实验组中,向0.2 mL PCR管中加入5 μL 3’-RACE cDNA,并向同一管中添加5 μL Master Mix。在空白对照组中,将5 μL PCR水加入 0.2 mL PCR管中,然后加入5 μL Master Mix。开始PCR程序(循环40次):初变性94 ℃1 min;变性94 ℃30 s;退火60 ℃30 s;延伸72 ℃3 min;终延伸72 ℃10 min。
凝胶电泳分析DNA产物:称量0.45 g的琼脂糖粉末,并向粉末中加入35 mL TBE溶液,放入微波炉中直到琼脂糖粉末溶解。冷却琼脂糖后,向琼脂糖中添加3.5 μL SYDR安全DNA凝胶,即将琼脂糖倒入凝胶盒中并插入合适的梳子。凝胶在室温下干燥需要40 min,凝胶干燥后用TBE溶液填充凝胶盒,加入体积为2.5 μL的DNA ladder。将体积为0.5 μL上样染料与1.5 μL RACE PCR样品混合,并倒入凝胶孔中,电泳在90 V下运行30 min,将凝胶转移至成像系统并进行分析。
DNA纯化:将PCR产物转移到1.5 mL管中,加入4倍体积的CP溶液,以分解琼脂糖并释放DNA和RNA。涡旋直至完全混合,将离心管涡旋直至完全混合,13 000×g下离心30 s,然后将液滴收集在盖中。将HiBind DNA柱插入2 mL管中。将从1.5 mL离心管收集的液滴添加到HiBind DNA柱中,丢弃废液,重复使用收集管。向离心管中加入700 μL的DNA洗涤液,13 000×g离心1 min,丢弃滤液,重复使用收集管。重复上述步骤以完全洗脱PCR产物中的任何杂质。将空的HiBind DNA柱13 000×g离心2 min,以干燥柱。将HiBind DNA柱转移到1.5 mL管中以提取DNA,向柱中加入体积为30 μL的PCR水,并在室温下储存2 min;13 000×g下离心1 min。
纯化的RACE-PCR产物与载体连接:将干燥的DNA样品用4 μL PCR水再悬浮,13 000×g离心30 s,并置于冰上。将pGEM-T载体13 000×g离心30 s,以收集管底部的产物。混合连接反应溶液体系:2×快速连接试剂2.5 μL,载体 (50 ng/μL)0.5 μL,PCR产物1.5 μL,T4 DNA连接酶(3 Weiss Unit/μL) 2.5 μL。将上述溶液混合并在室温下孵育1 h,然后在 4 ℃下过夜。
质粒导入:将含有连接反应的离心管离心,收集瓶底的内容物。在冰上将每个连接反应的2 μL内容物加入到1.5 mL离心管中。从-80 ℃冰箱中收集冷冻细胞,并将其放入冰盒中直至解冻。混合细胞后,将体积为50 μL的细胞转移到管中,离心管使之混合,并在冰上放置20 min。将离心管在42 ℃下加热47 s,并迅速放在冰上2 min。将体积为950 μL的SOC培养基加入到含有转化细胞的离心管中,在37 ℃下摇床(约150 r/min)培养3 h。将体积为120 μL的转化培养物转移到琼脂板上,将板在37 ℃下培养过夜。
蓝白筛选:将转化的板从培养箱中取出。用琼脂制备一个新的LB/氨苄青霉素/IPTG/X-Gal琼脂平板,并在平板底部画出曲线。接种环用本生灯火焰消毒。用冷接种环选择一个白色菌落,不接触其他细菌。在每个方格的琼脂上画出曲线。新的平板在37 ℃下培养过夜。
获取DNA质粒、扩增:将平板从培养箱中取出,取出白色菌落。准备好合适的0.5 mL离心管,每管加入20 μL水。用200 μL的枪头刮取白色菌落,将移液枪的枪头插入含有水的0.5 mL管中,使大肠杆菌细胞重新悬浮起来。所有离心管在100 ℃下加热5 min,并立即放在冰上5 min。将离心管13 000×g离心30 s,大肠杆菌细胞被溶解并储存于-20 ℃。将溶解的大肠杆菌细胞从冰箱中移出,将离心管涡旋30 s,
13 000×g离心5 min,分离出DNA质粒。为扩增反应准备扩增反应体系:5×克隆试剂10 μL,M13上游引物2.5 μL,M13下游引物2.5 μL,GoTaq聚合酶(5 U/μL) 0.25 μL,PCR水32.25 μL。M13上游引物:5’-GTA ACG CCA GGT TTC CCA G-3’;M13下游引物:5’-TGT GAG CGG ATA ACA ATT TCA C-3’。将47.5 μL的等量样品分装到离心管中,将2.5 μL的分离质粒加入离心管中,通过移液枪将样品混合,将离心管13 000×g离心30 s,收集离心管底部的质粒。启动PCR程序:初变性,95 ℃2 min;变性95 ℃30 s,退火55 ℃30 s,延伸72 ℃1 min,循环40次;终延伸72 ℃5 min。
DNA测序:凝胶电泳分析和克隆的PCR产物的纯化按上述方法进行。混合测序反应用的反应溶液体系:PCR水12.43 μL,M13上游引物(20 μmol/L)1.14 μL,反应混合试剂2.86 μL,循环测序试剂3.57 μL。将20 μL的母液加入0.2 mL离心管中,将2.5 μL的纯化DNA样品加入离心管中,混合,13 000×g离心30 s,收集底部产物。运行PCR程序:初变性96 ℃1 min;变性96 ℃20 s,退火55 ℃10 s,延伸60 ℃ 4 min,循环26次。PCR样品储存在冰箱里。醇沉并将体积为10 μL的样品转移到测序盘中,用自动测序仪测序。
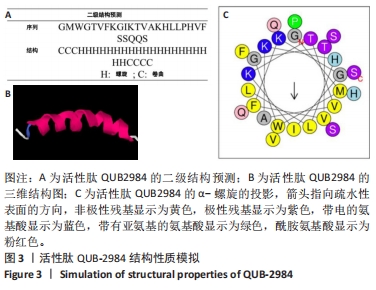
1.4.3 生物信息学工具模拟蛋白构效关系 应用生物大分子序列比对搜索工具的蛋白质BLAST程序,将核苷酸或蛋白质序列与序列数据库进行比较,以确定肽家族并预测成熟肽序列。应用I-TASSER网站预测蛋白质的二级结构和功能。应用HeliQuest网站来预测肽的疏水性、疏水性和净电荷。
1.4.4 固相合成法合成活性肽QUB2984 称量氨基酸(GMWGTVFKGIKTVAKHLLPHVFSSQQS),上样于多肽合成仪,并按照程序运行。通过裂解反应除去保护剂和与多肽相连的树脂。沉淀干燥后,向试管中加入5 mL溶液A(TFA∶水体积比=0.05∶99.95,) 加入溶液B(TFA∶水∶乙腈体积比=0.05∶29.95∶70.0)至30 mL以溶解肽。将试管置于冷冻干燥器中48-50 h。冻干肽被储存在-20 ℃的干燥器中。
1.4.5 纯化鉴定活性肽QUB2984
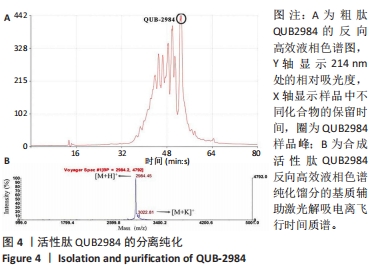
反向高效液相色谱法:使用高效液相色谱系统纯化多肽。固定相是C5分析柱,流动相为溶液A(三氟乙酸/水体积比=0.05/99.95)和溶液B(三氟乙酸/水/乙腈体积比=0.05/29.95/70.0)。用紫外检测器在214 nm处检测肽键。将10 mg合成肽溶解在含有1 mL溶液A和1 mL溶液B的试管中,涡旋并5 000×g离心10 min。溶液B以4 mL/min的流速洗涤20 min。然后用4 mL/min的溶液A和1 mL/min的溶液B以5 mL/min的流速平衡柱子20 min。将1mL溶解的肽溶液注入进样阀,流动相以5 mL/min的流速从80%的溶液A到100%的溶液B进行梯度洗脱,持续80 min。将每个峰的馏分收集,以便随后进行基质辅助激光解吸电离飞行时间质谱分析。
基质辅助激光解吸电离飞行时间质谱法:应用基质辅助激光解吸电离飞行时间质谱分析每个馏分中肽的分子质量,以确认分离的高效液相色谱馏分是否为目标肽。将每个高效液相色谱馏分2 μL 滴在干燥的板上,加入1 μL 30%的α-氰基-4-羟基肉桂酸,底物干燥后,将平板放在基质辅助激光解吸电离飞行时间质谱MS仪器上,用来鉴定纯化的肽。
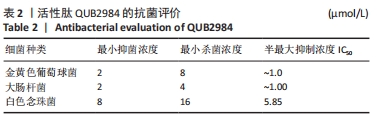
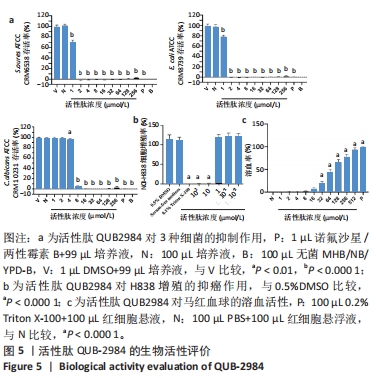
1.4.6 活性肽QUB2984的抗菌活性评价 革兰阳性细菌金黄色葡萄球菌(S. aureus)、革兰阴性细菌大肠杆菌(E. coli)和真菌致病性酵母白念珠菌(C. albicans)被用来评估活性肽的抗菌和杀菌活性。
细菌培养:将20 μL冻干的细菌在200 mL营养肉汤(NB)(大肠杆菌)/MHB培养基(金黄色葡萄球菌)/Y-PDB培养基(白色念珠菌)中进行培养。将烧杯在37 ℃(大肠杆菌、金黄色葡萄球菌)/26 ℃(白色念珠菌)的摇动培养箱中培养16-20 h。从3个培养瓶中各取20 mL悬浮液,放在37 ℃培养箱中过夜。
样品制备:将多肽溶于DMSO中,制成512×102 µmol/L的浓度。稀释得到一系列浓度的多肽溶液:512×102 µmol/L,256×102 µmol/L,128×102 µmol/L,64×102 µmol/L,32×102 µmol/L,16×102 µmol/L,8×102 µmol/L,4×102 µmol/L,2×102 µmol/L,1×102 µmol/L。
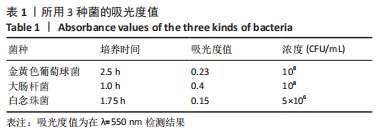
传代培养:将100 μL过夜生长的菌种转移到含温热营养肉汤的培养瓶中,并在摇动的培养箱中继续生长至对数期。达到表1中所示的吸光度值,说明菌落单位已达到预期值,可用于以下实验。孵化后,用新鲜的MHB培养基稀释细菌亚培养物,以确保5×105 CFU/mL的浓度。金黄色葡萄球菌和大肠杆菌以100 µL亚培养液和19.9 mL新鲜MHB/营养肉汤进行稀释。白色念珠菌以2 mL培养液和18 mL新鲜YPD-B培养基稀释。CFU按照如下公式计算:CFU=N/3×50×10n (N代表每个浓度下的细菌总量,n是稀释比例n=2,3,4)。
计数:将MHA/NA/Y-PDA管在沸水浴中融化,融化的MHA被转移到每个培养皿中。培养皿被轻轻旋转并干燥。对于每个细菌,需要使用离心管。将900 µL PBS与第一个离心管中的100 µL培养物混合,进行十倍稀释。然后将第一管中的100 µL溶液转移到第二管中,制成102稀释液。其余的以同样的方式进行,直至104稀释。将每支试管中的10 µL溶液滴到MHA平板上,并进行每个浓度的重复。培养皿培养一夜,对每个浓度梯度的菌落数量进行计数。用计数的平均值来计算亚培养液中的原始CFU,将其与应获得的适当CFU数进行比较,以确定细菌是否处于对数生长期,以用于抗菌活性评价。
加样:①实验组样品:1 µL活性肽肽溶液+99 µL细菌培养液;②阳性对照:1 µL诺氟沙星(商业渠道,英国)(2 mg/mL)+99 µL培养液(细菌)[终质量浓度:20 µg/mL]/1 µL两性霉素B(1 mg/mL)+99 µL培养液(酵母)[终浓度:10 µg/mL];③阴性对照:100 µL 细菌培养液;空白对照,100 µL无菌MHB/NB/YPD-B;④溶剂对照:1 µL DMSO+99 µL细菌培养液。实验组多肽浓度为512×102 µmol/L,256×102 µmol/L,128×
102 µmol/L,64×102 µmol/L,32×102 µmol/L,16×102 µmol/L,8×102 µmol/L,4×102 µmol/L,2×102 µmol/L,1×102 µmol/L。将所有样品和对照组排列在96孔板中,重复3次。将96孔板放在摇床上使混匀15 min,在37 ℃下培养过夜。
最小抑菌浓度检测:没有细菌生长的最低浓度是最小抑菌浓度。为了进一步测试细菌的存活率,打开96孔板的盖子,用多功能酶标仪在550 nm处测量每个孔的吸光度值。
细胞存活率(%) = (Asample-Ablank)/(AVehicle-Ablank) × 100% (1)
式中,ASample为实验样品孔的吸光度平均值,ABlank为不含细菌孔的溶液吸光度平均值,Avehicle为溶剂对照组孔的吸光度平均值。
最小杀菌浓度检测:最小杀菌浓度是杀死细菌的最低浓度。在多功能酶标仪检测后,将具有杀菌活性的最低浓度培养液加入96孔板的MHA/NA/Y-PDA培养皿中,每孔10 μL。干燥后,再将培养皿培养16-24 h,在培养皿表面观察细菌菌落,没有细菌存活的浓度是最小杀菌浓度。
1.4.7 活性肽QUB2984的细胞毒性评价 将人类非小细胞肺癌细胞系(H838)在RPMI-1640培养基中培养。所有的血清培养基含体积分数10%胎牛血清和体积分数1%抗生素。
MTT法检测细胞毒性:用胰蛋白酶消化NCI-H838细胞,13 000×g离心30 s,加入4 mL培养基。将50 µL细胞悬液和50 µL锥虫蓝加入到1.5 mL离心管中混匀。将50 µL的细胞悬液加入血细胞计数仪,在显微镜下观察并计算细胞数。细胞浓度用以下公式计算:细胞浓度(细胞/mL)=(计数的细胞总数/计数的大方格数)×稀释系数×104。用新培养基稀释细胞悬液,将细胞稀释到5 000个/100 µL的浓度,达到计算的浓度。然后将细胞悬液100 µL加入96孔板培养24 h,弃培养液,更换为100 µL新鲜无血清培养基,在37 ℃、体积分数5%CO2培养箱中培养4 h,饥饿细胞。将1 mg的纯冻干肽溶解在0.5%PBS和0.5%DMSO溶液中,制成最终浓度为10-2 mol/L的储备溶液。
实验分5组进行,将不同溶液加入96孔板每个孔中,对每个浓度的3个重复进行评估:多肽样品组加入4 µL肽(浓度从10-4 mol/L到10-9 mol/L)+396 µL细胞培养液;阳性对照组加入1 µL Triton X-100+999 µL细胞培养液;溶剂对照组加入2 µL DMSO+2 µL H2O+396 µL 细胞培养液;生长对照组加入400 μL细胞培养液;空白对照组加入400 μL细胞培养液,不加细胞与MTT。将96孔板在37 ℃、体积分数5%CO2培养箱内培养24 h。在黑暗环境下,除了空白对照,将MTT(5 mg/mL)加入96孔板孵化4-6 h,吸去溶液,加入100 µL DMSO,将平板在37 ℃的摇动培养箱中培养10 min,用酶标仪在λ=570 nm处测量吸光度值。细胞存活率用以下公式计算:
细胞存活率(%) = (Asample-Ablank)/(Agrowth-Ablank) × 100% (2)
式中,Asample为实验样品组的吸光度平均值,Agrowth为生长对照组孔的吸光度平均值,Ablank为空白对照组孔的吸光度平均值。
1.4.8 活性肽QUB2984对马红细胞溶血活性鉴定
马红细胞的预处理:将2 mL全马血液(商业渠道,英国)转移到50 mL离心管中,并加入30 mL PBS清洗血液。将血液930×g离心5 min,去除上清液,加入30 mL PBS;13 000×g离心30 s,直到上清液变清;清洗后加入50 mL PBS稀释红细胞,以制备体积分数4%的悬浮液。
多肽溶液的制备:称量冻干的多肽,溶解在PBS中,制备最终浓度为1 024 μmol/L的储备溶液。将原液稀释,得到一系列不同浓度的多肽溶液512,256,128,64,32,16,8,4,2,1 μmol/L,装在0.5 mL管中。
实验分组处理:实验分3组,每组包括3个重复,加入96孔板:实验组加入100 μL多肽溶液+ 100 μL红细胞悬浮液;阳性对照组加入100 μL 0.2% Triton X-100+100 μL红细胞悬浮液;阴性对照组加入100 μL PBS+100 μL红细胞悬浮液。准备好每组样品后,将所有试管与血液一起放入37 ℃的培养箱中培养2 h。孵化后取出试管,900×g离心5 min,从每支试管中取出100 μL的上清液,转移到96孔板中,用酶标仪在550 nm处测量每个孔的吸光度值,用GraphPad Prism软件计算溶血数据,公式如下:
溶血率(%)= (Asample-Ablank)/ (Anegative-Ablank)×100% (3)
式中,Asample为实验组吸光度平均值,Anegative为阴性对照组吸光度平均值,Apositive为阳性对照组吸光度平均值。
1.5 主要观察指标 活性肽QUB2984的结构和性质及生物活性。
1.6 统计学分析 抑菌实验利用GraphPad Prism 7.0软件进行分析,其中One-way ANOVA用于组间比较,图中的误差线代表标准误差。对显著性差异水平的表示为:P < 0.05,P < 0.01,P < 0.001,P < 0.000 1。抑癌实验:统计学意义P值通过GraphPad Prism软件进行单因素方差分析,以比较肽样品组的细胞活力值与生长对照组的细胞活力值。溶血实验数据由GraphPad Prism软件计算,采用单因素方差分析检验。