2.1 MITF的发现及结构 1942年柏林的Paula Hertwig在受辐射小鼠的后代中首次发现MITF位点的突变,突变体纯合子小鼠眼睛无色素、大小不一、胡须弯曲、缺失上切牙和下切牙口以及小鼠断奶后很快死亡。杂合子小鼠在出生时仅表现出减少的眼睛色素沉着[10,20-21]。Hertwig将新基因称为“Microphthalmus”(符号“m”)[22]。后来,在其他脊椎动物(人、果蝇、黑猩猩及猪)中也同样发现了该基因位点的许多其他突变等位基因。MITF是在一定的组织/细胞(包括破骨细胞)中表达的转录因子[23]。MITF具有一系列在其第一外显子和启动子方面不同的亚型。人MITF基因位于人3号染色体上,长为 228 902 bp,存在18个转录本,包含23个外显子。MITF基因不仅编码一种MITF蛋白,而且编码一个大家族的异构体,它们在序列、翻译后修饰和表达模式上都不相同。到目前为止,MITF不仅与自身形成二聚体,还与3个相关的合作伙伴TCFE3,TCFEB和TCFEC形成各自的复杂性[24]。此外,MITF可以与其他伴侣蛋白或辅因子相互作用,以及这些相互作用受到翻译后修饰(如磷酸化、SUMO化、泛素化、甲基化和乙酰化)的影响。MITF表达为一系列在其第一个外显子和启动子中的亚型。对于大多数亚型来说,最出的外显子是同种型特异性外显子,被剪接到外显子1B的后期部分,然后连接到外显子外部。目前,已在小鼠中鉴定出9种MITF亚型,即MITF-A,-B,-C,-D,-E,-H,-J,-M和-mc。MITF亚型以细胞类型特异性方式表达,其转录活性与靶基因重叠,但在MITF亚型间有明显差异。
人类中的杂合MITF突变导致神经性耳聋和色素沉着缺陷。常染色体显性遗传MITF突变与两种高度重叠的耳聋和色素沉着障碍相关:Waardenburg综合征2A型和Tietz综合征。先天性色素沉着缺陷和感觉神经性耳聋分别归因于MITF在皮肤和耳蜗血管纹中的黑素细胞的分化和存活。MITF的常染色体-隐性遗传或复合-杂合遗传以前在人类中尚未见报道。突变不会影响MITF与其他MIT家族转录因子的二聚化,而是改变同源二聚体和异源二聚体的核迁移和DNA结合,从而使突变体等位基因起主导作用。
2.2 MITF在破骨细胞中的研究进展
2.2.1 MITF促进破骨细胞的分化 骨是一种高度动态的组织,经历连续重塑,受各种因素的调节,包括细胞因子、趋化因子、激素和机械刺激。骨重塑依赖于成骨细胞和破骨细胞维持的骨形成和骨吸收两个动态过程间的平衡,若这2种力量之间的不平衡则会引起人类骨疾病的发展,包括骨质疏松症、骨硬化病、骨的佩吉特氏病、恶性骨疾病、类风湿和骨关节炎及根尖周炎等,其致病机制与破骨细胞产生过剩或者过度激活尤其相关。破骨细胞由肿瘤,炎症细胞因子介导,破骨细胞的前体是过度增殖和分化成为多核的造血干细胞及巨噬细胞等。
骨重塑是去除和修复受损骨骼的过程。通过这个过程,破骨细胞被逆转,然后被新的骨骼形成所取代。它发生在整个骨组织中。重塑的第一个重要步骤是核因子κB受体活化因子配体诱导的造血前体活性破骨细胞的产生。核因子κB受体活化因子配体的天然受体是骨保护素,也称为破骨细胞生成抑制因子,破骨细胞形成受破骨细胞抑制因子和核因子κB受体活化因子配体之间平衡的调节。破骨细胞是造血起源的多核细胞,其在骨量调节和骨质量方面起着关键作用。而骨骼对于破骨细胞的需要是在经受持续的机械变化和营养需求下,特别是在骨折愈合期间。
据报道,破骨细胞从骨折愈合的早期阶段起是活跃的,并且在整个过程中保持活跃,这些破骨细胞在单纯的吸收活性(去除多余的矿化骨痂组织)和主动的重塑作用之间有明显的改变,并与成骨细胞协同优化骨结构和排列。破骨细胞活性的状态取决于骨组织愈合阶段以及愈伤组织内细胞的定位。它表明破骨细胞在破骨阶段比平时有活跃。因此,研究抑制破骨细胞增殖及分化的靶蛋白对于治疗骨破坏相关疾病起到了重要的作用。
MITF由Luchin等在2000年建立并且第一次提出与破骨细胞的分化有关。MITF诱导造血干细胞分化为破骨细胞前体,是破骨细胞生成的关键转录因子[25]。已有研究表明,MITF是对包括破骨细胞、黑色素细胞、视网膜色素上皮细胞、肥大细胞和天然杀伤细胞在内的几种细胞谱系的正常发育所必需的。小鼠模型研究表明MITF在调节破骨细胞发育中起关键作用。不同有机体中MITF的突变导致破骨细胞发育缺陷,从而导致骨硬化的发生。MITF在破骨细胞前体的存活中起着非常重要的作用,并且涉及许多破骨细胞分化标志物的调节,骨髓谱系的造血前体的主要调节因子(PU.1),骨硬化病相关跨膜蛋白1(Ostm1)和破骨细胞相关受体[26-27]。遗传学证据表明,MITF在破骨细胞的分化过程中发挥了重要的作用[7]。由于缺乏成熟的功能性破骨细胞,MITF突变小鼠的骨骼显示出骨质疏松症的典型症状[28]。
在破骨细胞中,MITF已被证明可激活骨破坏标志性因子的表达。包括抗酒石酸酸性磷酸酶、组织蛋白酶K、破骨细胞相关受体和上皮性钙黏附蛋白[29]。抗酒石酸酸性磷酸酶是正常软骨矿化和成体骨重建所需的金属酶。组织蛋白酶K是一种半胱氨酸蛋白酶,对骨有机基质的降解非常重要。破骨细胞相关受体是破骨细胞中ECM胶原蛋白的受体,而且据报道指出破骨细胞相关受体受MITF和核因子激活的T细胞c1(NFATc1)的调节[14]。破骨细胞分化和成熟的调节不仅仅是由成骨细胞和基质细胞的分泌蛋白所决定。MITF是一种以组织特异性方式表达的转录因子。MITF也在单核细胞-巨噬细胞系和细胞中表达[30]。
MITF至少分两步调节破骨细胞分化。首先,MITF上调了与破骨细胞活性相关的基因的表达,如抗酒石酸酸性磷酸酶、氯离子通道蛋白7(CLCN7)、组织蛋白酶K和v-ATP酶d2等[31];其次,MITF通过细胞融合调节破骨细胞发育。树突状细胞特异性跨膜蛋白(DC-STAMP)是破骨细胞有效细胞-细胞融合所必需的,并且MITF可正确调节树突状细胞特异性跨膜蛋白的转录[32],见图1。MITF在刺激细胞融合中的活性已在不同细胞类型中也有表现。成肌细胞和新生肌管中的MITF表达是通过上调整合素α9(Itga9)的表达来实现肌管成熟的关键。
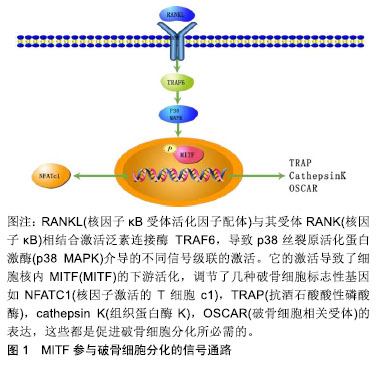
2.2.2 敲除MITF抑制破骨细胞的分化 MITF缺失后等位基因产生一种受损的骨表型,由于缺乏功能性破骨细胞,MITF敲除小鼠的骨骼出现骨质疏松症的迹象[21]。研究指出,MITF突变可以导致新生小鼠出现骨代谢的异常发育。MITF的非翻译区(UTR),因此导致MITF的降解和MITF的表达和转录活性的下降[33]。而且,MITF被确定为促进巨噬细胞的存活,同时MITFmi/mimice表现出了严重的骨硬化病。MITF-突变的蛋白质被募集到破骨细胞中的靶基因启动子,尽管效率比野生型MITF降低[34]。然而,这些MITF突变蛋白不能有效地将MITF的破骨细胞特异性共伴侣骨髓谱系的造血前体的主要调节因子(PU.1)募集到靶启动子。研究证明突变体MITF蛋白导致破骨细胞分化缺陷,因为它不能将来自核因子κB受体活化因子配体的信号传导整合到破骨细胞中的MITF靶基因转录[35]。此外,有研究表明miR-155抑制了MITF的表达,从而抑制了核因子κB受体活化因子配体诱导的破骨细胞分化。从而发现MITF敲除抑制了抗酒石酸酸性磷酸酶,降钙素受体,V-ATP酶d2和组织蛋白酶K的表达,这些都提示了MITF的敲除抑制了破骨细胞的分化。
为了确定MITF对破骨细胞分化的影响,有学者采用阴性对照siRNA(对照)或si-MITF转染骨髓巨噬细胞。检测破骨细胞分化标记基因包括(抗酒石酸酸性磷酸酶、降钙素受体、V-ATP酶d2和组织蛋白酶K)的相对表达水平,数据显示,用si-MITF转染的骨髓巨噬细胞中的相对TRAP表达低于用对照siRNA转染的骨髓巨噬细胞,说明MITF敲除抑制抗酒石酸酸性磷酸酶的表达;同样在降钙素受体、V-ATP酶d2和组织蛋白酶K组中观察到类似的结果,以上结果证明了敲除MITF会抑制破骨细胞的分化。其他因素也在破骨细胞生成中起重要作用。报道指出小鼠破骨细胞生成过程中特异性诱导了MITF的E亚型(Mitf-E),而不是A亚型(Mitf-A)的表达,这种诱导是有效形成破骨细胞所必需的。
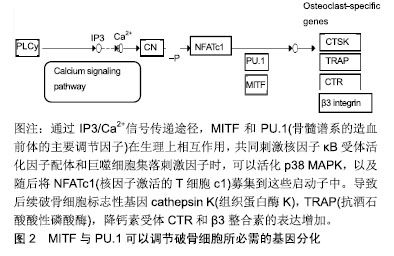
2.2.3 核因子κB受体活化因子配体通过MITF促进破骨细胞的分化 破骨细胞通过分解酸和各种酶降解骨基质,使骨再吸收细胞的过度形成和活化导致骨的吸收超过骨的形成,最终导致净骨丢失。破骨细胞通过单核细 胞-巨噬细胞谱系的前体细胞的融合而形成。破骨细胞分化主要由成骨细胞和基质细胞产生的分子诱导,其包括巨噬细胞集落刺激因子(M-CSF)和NF-κB配体的受体活化剂(RANKL)。研究表明核因子κB受体活化因子配体和巨噬细胞集落刺激因子均可以刺激破骨细胞分化所必须的转录因子表达增高,这些转录因子包括MITF,骨髓谱系的造血前体的主要调节因子(PU.1),c-Fos和核因子激活的T细胞c1(NFATc1)。这些因子作用于单核细胞/巨噬细胞谱系的髓样前体进而诱导其成为破骨细胞,并介导破骨细胞的信号通路。它们进一步激活了细胞-基质相互作用的特征程序,产生许多骨降解酶以产生高度酸化的微环境,可以分解骨基质下面的矿物质和有机成分。与其伴侣骨髓谱系的造血前体的主要调节因子(PU.1)一起被证明可以调节破骨细胞所必需的基因分化。在Sharma等发表研究中指出,MITF可以在刺激的破骨细胞前体中的酸性磷酸酶5和组织蛋白酶K启动子处检测到PU和PU.1。但是在只有巨噬细胞集落刺激因子的条件下,两种基因都不表达。在共刺激时用巨噬细胞集落刺激因子和核因子κB受体活化因子配体后,可以启动破骨细胞分化和激活这些启动子SWI/SNF染色质重塑的共同募集复合物,活化的p38 MAPK,以及随后将核因子激活的T细胞c1募集到这些启动子中。此外,转录生长因子β增强核因子κB受体活化因子配体诱导的Mitf-E表达和鼠破骨细胞形成。这些发现都表明Mitf-E和转录生长因子-beta是鼠细胞系统中破骨细胞生成的增强。已有研究表明,MITF是对包括破骨细胞,黑色素细胞,视网膜色素上皮细胞,肥大细胞和天然杀伤细胞在内的几种细胞谱系的正常发育所必需的[36]。小鼠模型研究表明MITF在调节破骨细胞发育中起关键作用。不同有机体中MITF的突变导致破骨细胞发育缺陷,从而导致骨硬化的发生。MITF与PU.1可以调节破骨细胞所必需的基因分化,见图2。
2.2.4 磷酸化p38通过MITF促进破骨细胞的分化 MITF已被证明是破骨细胞中巨噬细胞集落刺激因子和核因子κB受体活化因子配体信号通路的受体激活剂的靶点。核因子κB的受体活化剂位于破骨细胞前体细胞的细胞膜上。核因子κB受体活化因子配体和核因子κB结合诱导单核细胞发育成破骨细胞谱系。而核因子κB的激活导致下游信号转导的激活,核因子κB受体活化因子配 体-核因子κB的结合,其触发核因子κB和丝裂原活化蛋白激酶途径(JUN,ERK及p38)[37-39]。在破骨细胞发育过程中,破骨细胞被巨噬细胞集落刺激因子和核因子κB受体活化因子配体刺激,其启动一系列事件,导致通过 磷-ERK(pERK)和磷酸化p38(p-p38)激活MITF。有研究表明MITF复合物整合了在破骨细胞分化过程中的相关基因,如组织蛋白酶K和酸性磷酸酶5(Acid phosphatase 5,Acp5)适当时间表达所需的信号。核因子κB受体活化因子配体与其受体核因子κB的结合导致由前体细胞中c-Jun N末端激酶(c-Jun N-terminal Kinase,JNK)、p38 MAPK和细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)介导的不同信号级联的激活[40]。特别是,p38 MAPK的刺激对核因子κB受体活化因子配体诱导的破骨细胞分化至关重要,因为它调节了几种破骨细胞标志性基因的表达。它的激活导致了MITF的下游活化,而后者又控制编码酒石酸抗性酸性磷酸酶(TRAP,由Acp5编码)和组织蛋白酶K的基因的表 达[41]。因此,p38 MAPK的激活在核因子κB受体活化因子配体诱导的破骨细胞分化中起着重要的作用。这种刺激导致MITF的下游活化,这是破骨细胞分化和控制破骨细胞相关基因表达所必须的。特别是,MITF是一种破骨细胞分化的主要调节因子,通过与核因子激活的T细胞c1和c-Fos合作,而核因子κB受体活化因子配体通过调节许多破骨细胞特异性基因,如抗酒石酸酸性磷酸酶、组织蛋白酶K、降钙素受体、破骨细胞相关受体和核因子激活的T细胞c1诱导成熟的活性破骨细胞的形成[42]。MITF在破骨细胞分化中的重要作用已经通过对遗传修饰的突变小鼠进行的几项研究中得到了很好的证实。报告指出破骨细胞特异性条件性MITF缺陷小鼠由于破骨细胞生成受损而发生骨硬化。MITF作为核因子κB受体活化因子配体诱导的破骨细胞分化的关键转录因子提供了强有力的证据。MITF缺陷的胚胎干细胞不能在核因子κB受体活化因子配体的作用下分化成破骨细胞[43]。组成型活性MITF在骨髓单核细胞/巨噬细胞中的过表达导致前体细胞,即使在没有核因子κB受体活化因子配体的情况下也能进行有效分化,尽管没有核因子κB受体活化因子配体,MITF也可以在破骨细胞前体细胞中的异位表达,在这些细胞中诱导破骨细胞分化。这些结果表明MITF是体外和体内破骨细胞分化不可缺少的因素。因此,了解破骨细胞中MITF调节的分子机制可以为与过度破骨细胞分化和功能相关的骨疾病提供新的治疗策略。
2.2.5 组蛋白脱乙酰基酶7与MITF相结合MITF抑制破骨细胞的分化 适当的骨骼稳态或病理性骨质流失之间的平衡需要调节破骨细胞的形成和功能。骨质疏松症等疾病,转移性骨病和关节炎都涉及过度的破骨细胞活性,导致骨质破坏。研究表明破骨细胞分化由组蛋白脱乙酰基酶7调节,作为MITF的阻遏物是破骨细胞分化所需的。因此,在破骨细胞谱系中缺失组蛋白脱乙酰基酶7,会导致破骨细胞的生成量显著减少。MITF作为破骨细胞调节剂的重要性早已得到了重视研究表明,组蛋白脱乙酰基酶7参与了对MITF下游标志因子的作用,如组织蛋白酶K、核因子激活的T细胞c1、树突状细胞特异性跨膜蛋白和Atp6v0d2蛋白抗体(Anti-ATP6V0D2)。在进一步的巨噬细胞集落刺激因子和核因子κB受体活化因子配体刺激的分化期间,通过ERK1/2和p38 MAPK磷酸化MITF,导致共激活蛋白的募集和转录激活。结果显示,MITF被巨噬细胞集落刺激因子磷酸化,通过丝氨酸73上的ERK1/2和核因子κB受体活化因子配体,通过丝氨酸307上的p38 MAPK。磷酸化MITF增强其激活酸性磷酸酶5,破骨细胞相关受体和树突状细胞特异性跨膜蛋白启动子的能力通过募集转录共激活蛋白复合物。通过短发夹RNA(Short hairpin RNA,shRNA)敲低组蛋白脱乙酰基酶7的表达会导致增强巨噬细胞集落刺激因子/核因子κB受体活化因子配体在体外刺激破骨细胞生成。而且,MITF/组蛋白脱乙酰基酶7的互动被核因子κB受体活化因子配体刺激破坏,表明组蛋白脱乙酰基酶7的抑制作用减弱了MITF可能有助于核因子κB受体活化因子配体诱导的基因表达和破骨细胞分化。Jin证实了组蛋白脱乙酰基酶7作为破骨细胞的抑制剂,通过改变核因子激活的T细胞c1,β-连环蛋白和细胞周期蛋白D1,进一步抑制了破骨细胞的增殖及分化。但是关于MITF和组蛋白脱乙酰基酶共同阻遏物之间的相互作用,目前的研究较少。组蛋白去乙酰化酶抑制剂已被证明具有抑制破骨细胞分化,表明组蛋白脱乙酰基酶的缺失可能会减少破骨细胞形成和增加骨量。实际上,组蛋白脱乙酰基酶抑制剂正在作为炎症性骨细胞病症如关节炎和牙周炎的治疗方法的研究。研究表明,组蛋白脱乙酰基酶3的缺失减少了破骨细胞的形成。然而,组蛋白脱乙酰基酶7的抑制增强了巨噬细胞集落刺激因子和核因子κB受体活化因子配体诱导的破骨细胞增殖及分化,组蛋白脱乙酰基酶7是破骨细胞形成的负调节剂。组蛋白脱乙酰基酶7过表达阻断了破骨细胞生成在体外,并显示组蛋白脱乙酰基酶7的条件性敲除降低了体内骨量由于核因子激活的T细胞c1的错误调节导致破骨细胞骨吸收增加,β-连环蛋白和细胞周期蛋白D1,但是组蛋白脱乙酰基酶的抑制作用是对骨骼健康是有害的。实际上,文献中有一些报告表明长期给药组蛋白脱乙酰基酶抑制剂丙戊酸作为癫痫患者的抗惊厥药后骨折的风险增加,骨密度降低。

.jpg)
.jpg)