Chinese Journal of Tissue Engineering Research ›› 2023, Vol. 27 ›› Issue (17): 2699-2707.doi: 10.12307/2023.477
Macrophage polarization and periodontitis
Yuan Shuyue1, Liu Chunyan2, Liu Bing3, Zhao Fei3
- 1Hebei Key Laboratory of Stomatology, Hebei Clinical Research Center for Oral Diseases, School and Hospital of Stomatology, Hebei Medical University, Shijiazhuang 050017, Hebei Province, China; 2Department of Orthodontics, 3Department of Periodontics, Hebei Key Laboratory of Stomatology, Hebei Clinical Research Center for Oral Diseases, School and Hospital of Stomatology, Hebei Medical University, Shijiazhuang 050017, Hebei Province, China
-
Received:2022-06-22Accepted:2022-08-19Online:2023-06-18Published:2022-10-25 -
Contact:Liu Bing, Master, Chief physician, Associate professor, Department of Periodontics, Hebei Key Laboratory of Stomatology, Hebei Clinical Research Center for Oral Diseases, School and Hospital of Stomatology, Hebei Medical University, Shijiazhuang 050017, Hebei Province, China -
About author:Yuan Shuyue, Master candidate, Hebei Key Laboratory of Stomatology, Hebei Clinical Research Center for Oral Diseases, School and Hospital of Stomatology, Hebei Medical University, Shijiazhuang 050017, Hebei Province, China -
Supported by:2021 Government-Funded Medical Excellent Talent Project of Hebei Province, No. 361029 (to LCY); 2022 Hebei Provincial Medical Research Project, No. 20221454 (to ZF); 2019 Government-Funded Clinical Medical Talents Training and Basic Research Project of Hebei Province, No. 361029(MXZB00271) (to LB)
CLC Number:
Cite this article
Yuan Shuyue, Liu Chunyan, Liu Bing, Zhao Fei. Macrophage polarization and periodontitis[J]. Chinese Journal of Tissue Engineering Research, 2023, 27(17): 2699-2707.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
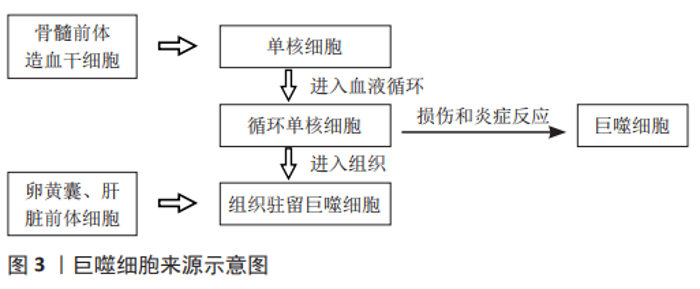
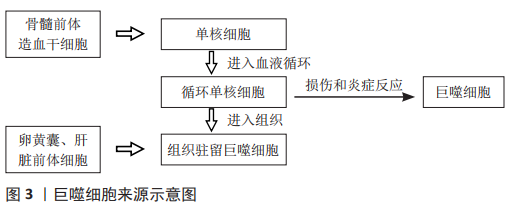
2.1 巨噬细胞 2.1.1 巨噬细胞的来源 长期以来,人们普遍认为巨噬细胞来源于循环单核细胞,循环单核细胞是由起源于骨髓前体造血干细胞的单核细胞在骨髓中成熟分化后进入血液循环形成的。当受到某些刺激时,循环单核细胞可穿破血管进入各组织中,分化为巨噬细胞[5-6]。研究发现,循环单核细胞可在损伤或炎症反应中分化成熟为巨噬细胞,或通过迁移到组织中并成为巨噬细胞[3], 即组织驻留巨噬细胞。但组织驻留巨噬细胞并非均由循环单核细胞发育而来,在对小鼠遗传谱系追踪研究中发现,在许多组织中,巨噬细胞起源于卵黄囊或胎肝的前体细胞。这些组织常驻巨噬细胞具有自我更新能力且存活时间长,能够在无单核循环细胞存在的情况下维持稳态[5-7],但是这两种来源的巨噬细胞对组织稳态的作用仍然未知,需要进一步研究[5]。巨噬细胞来源示意图,见图3。"
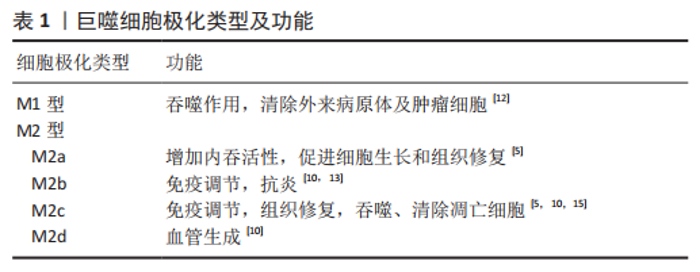
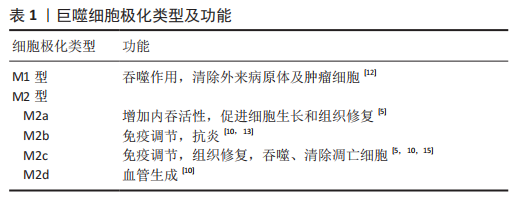
2.1.2 巨噬细胞极化类型及功能 巨噬细胞具有高度的可塑性,在不同环境及各种刺激下可以极化为不同的表型进而发挥不同的功能,主要为M1促炎表型和M2抗炎表型[8-9]。M2型巨噬细胞还可以进一步分为以下表型:M2a,M2b,M2c和M2d[10]。巨噬细胞极化是一个动态过程,在一定条件下M1,M2之间可以相互转化[11],这种转化使巨噬细胞在微环境变化时仍能维持组织稳态与机体平衡[5]。 M1型巨噬细胞具有强大的抗微生物活性和抗肿瘤活性,同时介导活性氧(ROS)引起组织损伤,损害组织再生和伤口愈合;具有较强的抗原递呈能力,能促进辅助性T细胞1(Th1)和辅助性T细胞17(Th17)免疫应答,在炎症早期促进炎症反应,杀伤细胞内感染的病原体。M2型巨噬细胞具有强大的吞噬能力、清除碎片和凋亡细胞、促进组织修复和伤口愈合,并具有促血管生成和促纤维化特性[4,9,12]。其中,M2a巨噬细胞,也称为伤口愈合巨噬细胞,可增加内吞活性,对寄生虫、生长刺激、组织修复、胶原蛋白形成和辅助性T细胞2、嗜碱性粒细胞和嗜酸性粒细胞的募集具有免疫力;M2b巨噬细胞,也称为调节性巨噬细胞,具有强大的抗炎和免疫抑制作用;M2c巨噬细胞,也称为灭活巨噬细胞,除具有发挥免疫抑制、促进血管生成、胶原蛋白形成等作用,还可以吞噬和清除死亡的细胞;M2d巨噬细胞也称为肿瘤相关巨噬细胞(TAM),功能是促进血管生成和肿瘤进展[12-15],见表1。"
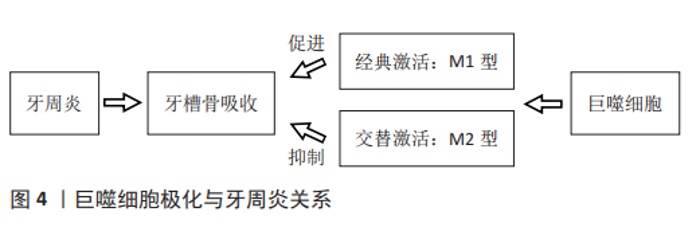
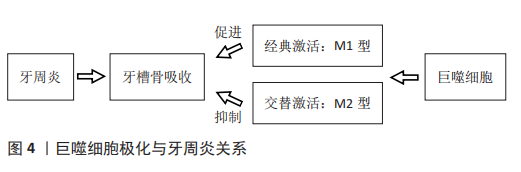
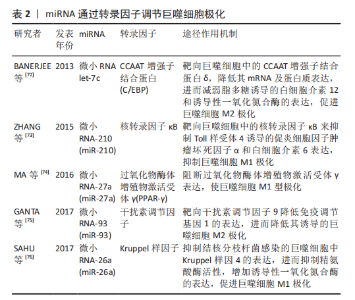
2.2 巨噬细胞极化在牙周炎中的作用 牙槽骨吸收是牙周炎进展标志,是导致牙齿脱落的原因。破骨细胞作为主要的骨吸收细胞是牙槽骨吸收所必需的,可在关键细胞因子巨噬细胞集落刺激因子、核转录因子κB配体(receptor activator of nuclear factor-κB ligand,RANKL)和骨保护素的调控下,从单核/巨噬细胞前体分化出来[2,16]。研究表明,M1型巨噬细胞通过吞噬作用促进病原体消除,刺激多形核中性粒细胞和T细胞的活化,并产生促炎细胞因子,如白细胞介素1β、白细胞介素6和肿瘤坏死因子α [17],且M1型巨噬细胞通过分泌促进辅助性T细胞1或辅助性T细胞17反应和刺激前破骨细胞的细胞因子使破骨细胞活化,导致RANKL受体激活剂的表达和骨破坏的升高[18-19],而M2型巨噬细胞通过分泌抗炎介质,如白细胞介素1受体拮抗剂(IL-1RA),参与炎症消退和组织再生[20]。因此,巨噬细胞可通过极化影响骨吸收与骨形成参与牙周炎疾病过程。牙周炎与巨噬细胞极化关系见图4。"
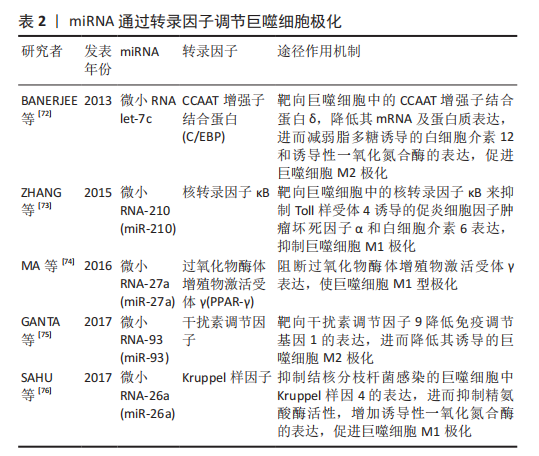
2.2.1 牙周炎中巨噬细胞极化类型 在牙周组织中,巨噬细胞向M1型极化主要是由辅助性T细胞1产生的干扰素γ和脂多糖主导的微生物相关因子诱导的,牙周病原菌通过脂多糖刺激巨噬细胞表面的CD14、Toll样受体和NOD样受体,诱导巨噬细胞分化为M1型。同时,牙周组织中巨噬细胞在辅助性T细胞2相关细胞因子白细胞介素4和白细胞介素13的刺激下,可以分化成M2型巨噬细胞,其在缓解炎症和组织修复方面发挥作用,特点是产生白细胞介素10并降低白细胞介素6的表达[21-22]。巨噬细胞在牙周疾病中促进组织分解,在牙周炎组织中观察到与巨噬细胞相关的更高水平的RANKL蛋白的受体激活剂[23]。研究表明,M1巨噬细胞是牙周炎的主要形式,且M1巨噬细胞可能通过诱导RANKL而转化为破骨细胞[24]。牙龈卟啉单胞菌(Pg)是一种关键的牙周病原体,可促进宿主和菌斑相关细菌之间的生态失调,从而导致牙周病的发病和进展[25]。牙龈卟啉单胞菌的脂多糖可通过Toll样受体2或Toll样受体4途径激活巨噬细胞,牙龈卟啉单胞菌的脂多糖激活Toll样受体2触发下游核转录因子κB刺激,增加M1型巨噬细胞促炎细胞因子的产生[26]。在白细胞介素1和肿瘤坏死因子α存在的情况下,牙龈卟啉单胞菌的脂多糖可诱导培养的人成纤维细胞和上皮细胞释放一种与牙周炎骨吸收相关的因子,即前列腺素E2(PGE2),促进促炎型M1样巨噬细胞极化[27]。 2.2.2 巨噬细胞极化与牙周炎牙槽骨吸收 上述研究发现牙龈卟啉单胞菌的脂多糖在白细胞介素1和肿瘤坏死因子α存在的情况下,巨噬细胞发生M1极化,不仅增强炎症反应,而且促进骨吸收,加重牙周炎破坏[25]。M1型巨噬细胞分泌促炎因子如白细胞介素1β、白细胞介素6、白细胞介素12等通过不同途径导致牙槽骨吸收[28]。有实验表明,牙龈卟啉单胞菌导致的老鼠牙周炎骨吸收模型中M1型巨噬细胞及其分泌的炎症因子明显较M2型增多,通过皮下注射双膦酸盐-脂质体后,M1型巨噬细胞及骨吸收明显减少[29],说明在牙周炎骨破坏过程中M1型巨噬细胞发挥主要作用。在牙龈卟啉单胞菌和结扎丝诱导的小鼠牙周炎模型中,用CC基序趋化因子配体2释放微粒处理组的小鼠牙槽骨吸收较未处理组减轻,且牙周炎模型中M2型巨噬细胞的数量增加[30],说明牙周炎过程中M2型巨噬细胞可减轻炎症反应,减少骨吸收。此外,在正畸骨吸收的研究中,M1/M2比例增加可导致牙槽骨的吸收,而M1/M2比例降低可抑制骨的吸收,即M1型巨噬细胞促进骨吸收[31-32]。 2.3 巨噬细胞极化在牙周炎发病中的机制 2.3.1 相关信号通路机制 JAK/STAT信号通路:Janus 激酶/信号转导和转录激活因子[Janus Kinase (JAK)-signal transducer and activator of transcription (STAT),JAK-STAT]信号通路与炎症和自身免疫性疾病的发病机制有关。许多参与自身免疫性疾病和炎症性疾病发病的机制涉及JAK和STAT细胞信号转导[33]。JAK家族包括JAK1,JAK2,JAK3和TYK24种成员,属于非受体蛋白酪氨酸激酶。JAK激活的STAT蛋白家族成员是调控巨噬细胞M1/M2极化的关键转录因子[34]。与牙周炎相关信号因子包括干扰素γ、肿瘤坏死因子α、白细胞介素1、白细胞介素4、白细胞介素6和白细胞介素10[35]。现已发现的JAK和STAT家族中,与巨噬细胞极化的相关成员有STAT-1,STAT-3和STAT-6。STAT-1和STAT-3/STAT-6的激活可调节巨噬细胞的极化和活性[4]。核转录因子κB和STAT-1激活促进巨噬细胞向M1表型极化,而STAT-3/STAT-6激活促进巨噬细胞向M2型极化[4]。 脂多糖诱导Toll样受体4通路激活JAK2/STAT1使巨噬细胞发生M1型极化[36]。白细胞介素10通过诱导p50-核转录因子κB二聚体、c-Maf和STAT3活性促进巨噬细胞M2型极化[37]。在牙周炎动物模型中发现STAT-3和STAT-5激活[38],因此,在脂多糖诱导牙周炎过程中巨噬细胞极化可能通过该信号通路影响牙周炎进展。细胞因子信号抑制因子(SOCS)是JAK/STAT通路的可诱导内源性调节因子,细胞因子信号抑制因子1和细胞因子信号抑制因子3分别是STAT1和STAT3的负反馈调节因子,可以通过不同的机制抑制JAK活性。研究表明细胞因子信号抑制因子3和STAT3的物理相互作用与脂多糖刺激的巨噬细胞中STAT3的激活状态之间存在反比关系,因此协调这二者的关系对于使用可以模拟细胞因子信号抑制因子3与STAT3物理相互作用的修饰肽调节炎症具有重要意义[39]。 一项关于Exendin-4(胰高血糖素样肽1受体激动剂)对骨质疏松大鼠骨形成影响的研究指出,Exendin-4通过促进巨噬细胞M2极化来预防骨丢失和促进骨形成,其通过胰高血糖素样肽1受体信号和STAT3信号之间的信号交叉,诱导骨髓巨噬细胞极化为M2型,促进骨髓间充质干细胞向吸收部位迁移,M2巨噬细胞又分泌转化生长因子β1来促进成骨[40-41]。以上研究表明在牙周炎过程中JAK/STAT信号通路可能通过影响巨噬细胞极化及成骨来调节炎症进展。 TLRs/核转录因子κB信号通路:研究证实Toll样受体(TLRs)能够识别病原相关分子模式,不同的Toll样受体能够识别不同的信号分子,进而作用于核转录因子κB,从而调节巨噬细胞对炎症细胞因子、感染和应激信号的激活[42]。Toll样受体-配体相互作用通过衔接蛋白MyD88和/或含有Toll/白细胞介素1受体结构域的衔接子诱导干扰素β(Trif)触发细胞内信号传导事件导致巨噬细胞中急性炎症反应快速激活。急性巨噬细胞活化是由炎症细胞因子受体介导,如肿瘤坏死因子受体[43]。牙龈卟啉单胞菌的脂多糖作为公认的引起牙槽骨吸收的关键因子,可以通过作为病原相关分子模式,被宿主体内模式识别受体如Toll样受体2或Toll样受体4等识别[44],激活核转录因子κB途径,诱导巨噬细胞向M1型极化。核转录因子κB途径可促进破骨细胞活化和分化,并产生白细胞介素1β、白细胞介素6和白细胞介素12等促炎因子,导致牙周组织破坏[28,45]。 PI3K/Akt信号通路:磷脂酰肌醇-3-激酶(phosphoinositide 3-kinase,PI3K)/蛋白激酶B(Akt)信号通路在巨噬细胞的存活、增殖和迁移中起着至关重要的作用,最近的研究表明 PI3K/Akt 信号传导是巨噬细胞极化和存活的关键调节剂[46]。PI3K/Akt通路被认为是巨噬细胞中Toll样受体和核转录因子κB信号传导的负调节剂[42,47]。研究发现,巨噬细胞中Akt活性的抑制有利于M1极化,PI3K活化和Akt的激活有利于白细胞介素4诱导的M1型巨噬细胞活化[4]。ARRANZ等[48]研究证明了Akt在巨噬细胞极化中发挥重要作用:Akt1缺乏会诱导M1型巨噬细胞,而Akt2的缺失会诱导M2型巨噬细胞。BAO等[49]研究发现,miR-32通过下调同源性磷酸酶张力蛋白激活PI3K/AKT信号通路,从而诱导M2型巨噬细胞极化。转化生长因子β、白细胞介素10、骨形态发生蛋白7等通过PI3K/Akt信号促进M2型巨噬细胞极化[42,50]。 另有研究发现PI3K/Akt信号通路阻断后,肿瘤坏死因子α介导B淋巴细胞诱导的成熟蛋白1(Blimp1)表达促进RANKL诱导破骨细胞生成引起牙槽骨吸收的作用也被阻断[51]。有研究表明,PI3K/Akt通路还影响巨噬细胞的代谢进程,糖原合成酶激酶3β(GSK3β)是一种重要的PI3K/AKT信号传导底物,对线粒体活动具有调节功能[52],而线粒体氧化磷酸化(OXPHOS)与巨噬细胞M2样极化表型有关[53],巨噬细胞清除受体1(MSR1)即CD206介导的PI3K/AKT/糖原合成酶激酶3β/β-连环蛋白信号增强了骨髓源性巨噬细胞共培养系统中成骨分化能力。此外增殖物激活受体γ共激活因子1α(PGC1α)作为共培养系统中巨噬细胞清除受体1激活的PI3K/AKT/糖原合成酶激酶3β/β-连环蛋白通路的靶基因,通过增强线粒体氧化磷酸化促进巨噬细胞M2型极化[52]。因此,PI3K/AKT通路可通过影响巨噬细胞极化及成骨与破骨来调控牙周炎的炎症进程。 MAPK信号通路:丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)是一种广泛存在于胞浆内的丝氨酸/苏氨酸蛋白激酶,在细胞增殖、分化、凋亡及存活等生理病理功能活动中具有重要作用。MAPK由4个信号通路组成,包括:p38丝裂原活化蛋白激酶(p38MAPK)通路、通路细胞外信号调节蛋白激酶1/2(ERK1/2)通路、c-Jun氨基末端激酶(JNK/SAPK)通路和细胞外信号调节蛋白激酶5(ERK5)/大丝裂原活化蛋白激酶1(BMK1)通路[54]。研究表明,牙周炎过程中释放的相关炎症因子可通过MAPK信号通路放大炎症反应[55]。 研究表明,p38,JNK,ERK信号通路在巨噬细胞向破骨细胞转化的各个环节中都发挥着重要作用,主要介导RANKL对破骨细胞的成熟与活化,导致牙槽骨吸收破坏[55]。研究发现牙周炎中神经生长导向因2(SLIT2)的过度表达可加剧牙周炎症使破骨细胞表达增加,且增强巨噬细胞的浸润和 M1巨噬细胞极化,上述作用是通过MAPK信号通路实现[56-57]。此外,M1型巨噬细胞也可以通过单独激活 MAPK信号通路来极化[58]。 Notch信号通路:研究表明,Notch信号通路可促进炎症进展,并可在炎症环境中促进巨噬细胞的促炎反应[42,59]。研究表明Delta样配体4(DLL4)/Notch1轴在促进巨噬细胞M1型极化方面起关键作用,同时阻断巨噬细胞M2型极化及其细胞因子表达[60]。Notch信号传导调节破骨细胞生成、RANKL和巨噬细胞集落刺激因子(M-CSF)以及细胞表面受体DAP12和FcRγ的激活,进而使巨噬细胞前体分化为破骨细胞[61]。因此,在牙周炎过程中,通过此通路可调节巨噬细胞极化类型,使M1/M2比例发生变化,影响牙周炎进展。 干扰素调节因子信号通路:干扰素调节因子是一个在巨噬细胞中表达的转录因子家族,根据该家族的成员,在巨噬细胞极化为M1或M2表型中发挥作用。炎症刺激可诱导干扰素调节因子5的表达,上调促炎症基因并下调M2型巨噬细胞相关基因的表达[62]。研究发现破骨细胞生成的关键调节分子干扰素调节因子8在减弱脂多糖诱导的炎症性骨吸收以及脂多糖和肿瘤坏死因子α诱导的破骨细胞生成方面发挥了重要作用[63]。转录因子RBP-J是破骨细胞生成的关键和中枢负调节因子,通过抑制c-Fos的表达和增加干扰素调节因子8的表达来抑制破骨细胞生成,减轻牙周炎症反应[64]。在牙龈卟啉单胞菌诱导的牙周炎中,干扰素调节因子3可以减少骨髓巨噬细胞中肿瘤坏死因子α的表达,说明干扰素调节因子3可减少牙周炎过程中巨噬细胞炎症因子释放[65],使M1型巨噬细胞表达减少,进而减轻牙周炎症。综上,干扰素调节因子可影响巨噬细胞极化表型及炎症因子的表达,从而在牙周炎中发挥作用。 2.3.2 表观遗传学机制 表观遗传是指DNA序列不存在改变,而基因表达发生可遗传的改变,其调控基因表达变化的机制主要涉及非编码RNA、DNA甲基化及组蛋白修饰。 非编码RNA:非编码RNA是一类调节性RNA,研究发现,非编码RNA可通过调控巨噬细胞极化通路中的关键蛋白而影响巨噬细胞极化状态,介导多种疾病的发生发展。已知非编码RNA如miRNA、长链非编码RNA(long noncoding RNA,lncRNA)和环状RNA(circRNA)在调控巨噬细胞极化中发挥重要作用[66]。 其中与M1型巨噬细胞极化相关的包括miR-27a,miR-223,let-7d-3p及miR-142-3p;与M2型巨噬细胞极化的有miR-181,miR-93,miR-1246及miR-26a。研究发现,miR-142-3p可抑制巨噬细胞M1型极化,并抑制其转化为破骨细胞来减少牙槽骨吸收[67]。LncRNA可调节免疫系统,研究表明LncRNA Nron(活化T细胞核因子抑制物[NFAT])与破骨细胞分化密切相关,在转基因小鼠实验中发现,Nron能显著抑制牙周炎破骨细胞的生成,在牙周炎治疗中显示出良好的转化应用潜力[68]。Nron主要促进核转录因子κB抑制因子(NKRF)向细胞核的移位,以抑制活化T细胞核因子1(Nfatc1)的转录,抑制牙周炎破骨细胞的生成。 lncRNA-Cox2可调节巨噬细胞极化,在体外,lncRNA-Cox2的敲低抑制了脂多糖诱导的炎症和M1巨噬细胞标志物的表达[69]。LncRNA Ptprj-as1在富含巨噬细胞的组织中高表达,并由促炎因子(如脂多糖)瞬时诱导[70],那么有理由怀疑LncRNA可能通过影响巨噬细胞极化影响牙周炎。circ_0085289通过调节let-7f-5p/SOCS6轴减轻了脂多糖刺激引起的牙周膜细胞(PDLC)损伤,这表明它是治疗牙周炎潜在生物标志物[71]。circRNA-003780、circRNA-010056及circRNA-010231在M1型巨噬细胞中高表达,且高表达的某些circRNA与miRNA存在相互作用,说明二者之间存在关系来调控巨噬细胞极化[66]。总之,这些非编码RNA在巨噬细胞极化与牙周炎关系中的具体机制尚未明确。此外,研究表明miRNA可通过调节某些转录因子,如过氧化物酶体增殖物激活受体、信号转导和转录激活因子、CCAAT增强子结合蛋白、干扰素调节因子、Kruppel样因子、GATA结合蛋白3、核转录因子κB 和c-MYC来调节巨噬细胞极化[72-76],有关miRNA调控巨噬细胞极化的相关研究见表2。 DNA甲基化:DNA甲基化是一种著名的表观遗传现象,已被证明是生物过程的重要调控因子。DNA甲基化通常发生在胞嘧啶-磷酸-鸟嘌呤(CpG)二核苷酸的胞嘧啶5’位置,并通过DNA甲基转移酶(DNMTs)的催化完成[77],包括DNA甲基转移酶1(DNMT1)、DNA甲基转移酶3a(DNMT3a)和DNA甲基转移酶3b(DNMT3b)。当甲基化发生时,DNA序列呈现出更集中的DNA结构,阻止转录因子进入启动子区域,导致基因失活[78]。 研究表明,DNA甲基化对牙周炎产生病理影响,且从牙周炎患者炎症组织中分离出来的基因具有不同的甲基化模式,研究认为这些基因是脂多糖介导的信号传导的调节因子[79-80]。目前研究称,DNA甲基化模式大多数与巨噬细胞的生理功能密切相关,或与调控巨噬细胞的各种信号通路中的因子相互作用。研究发现,敲除DNA甲基转移酶3b可促进巨噬细胞极化为M2表型,并抑制巨噬细胞的炎症反应[81]。在体外慢性牙周炎的模拟实验中发现,基质金属蛋白酶2相关基因的上调,影响巨噬细胞M1/M2极化。在人基质金属蛋白酶2处理的巨噬细胞中,诱导型一氧化氮合酶(iNOS为M1标记物)表达显著增加,而精氨酸酶1(Arg1为M2标记物)表达下降,即向M1巨噬细胞极化[77,82]。 一项关于肥胖个体DNA甲基化对巨噬细胞极化及炎症作用的研究发现,DNA甲基转移酶1的DNA甲基化在巨噬细胞极化和炎症的调节中起重要作用。髓样DNA甲基转移酶1缺乏会导致控制巨噬细胞替代激活的关键转录因子过氧化物酶体增殖物激活受体γ1DNA甲基化减少,而肥胖相关因子(如促炎细胞因子)显着增强了巨噬细胞中DNA甲基转移酶1介导的过氧化物酶体增殖物激活受体γ1启动子DNA甲基化,因此肥胖者过氧化物酶体增殖物激活受体γ1启动子的DNA甲基化失调,可能是对炎症刺激的反应,导致巨噬细胞极化失调[83]。Kruppel样转录因子是控制巨噬细胞极化的分子开关,研究称,Kruppel样转录因子通过调节miRNA和DNA甲基化在表观遗传学上参与巨噬细胞极化[84]。此外,研究发现,牙龈卟啉单胞菌的脂多糖可能诱导牙周膜细胞中DNA甲基转移酶1的高表达,引起Runt相关转录因子2(RUNX2)的高甲基化和Runt相关转录因子2的表达下调,从而抑制细胞从牙周膜向成骨细胞的分化[85]。 组蛋白修饰:组蛋白修饰包括甲基化、乙酰化、磷酸化、泛素化和类泛素化,这些变化通常参与建立与染色质状态改变相关的基因失调模式,从而导致许多疾病中的基因激活和基因沉默。编码酶的多个基因的表达负责催化和修饰各种组蛋白的翻译后修饰,例如甲基转移酶、去甲基化酶、乙酰转移酶和去乙酰化酶,这些酶在巨噬细胞M1/M2状态下差异表达。新发现表明,在炎症和免疫调节中,组蛋白修饰存在新的基于表观遗传的调节巨噬细胞极化机制[84]。 有研究表明,组蛋白去乙酰化酶3去除小鼠的巨噬细胞表现出强的白细胞介素4介导的替代激活,导致巨噬细胞M2极化[86]。 含Jumonji结构域-3(JMJD3)是巨噬细胞M2极化所必需的JmjC家族组蛋白去甲基化酶,脂联素(APN)是脂肪组织分泌的一种因子,据报道,在肥胖条件下的牙周炎中脂联素可能通过JMJD3-IRF4轴表观遗传调控巨噬细胞M2极化,进而改善牙周骨吸收[87]。也就是说组蛋白修饰参与牙周炎过程中巨噬细胞极化,影响牙周炎骨吸收。组蛋白乙酰转移酶(HA-Ts)和组蛋白去乙酰化酶分别介导组蛋白赖氨酸的乙酰化和去乙酰化。发现组蛋白H3和H4乙酰化与人单核细胞的发育阶段密切相关,并调节肿瘤坏死因子α启动子组蛋白去乙酰化酶功能的丧失同时对几种炎症性疾病有抑制作用。由干扰素γ极化的单核细胞通过ERK和p38 MAPKs信号通路增加肿瘤坏死因子α启动子处的组蛋白H4乙酰化,导致转录因子2(TF-2)激活和巨噬细胞M1极化[88]。 促炎细胞因子(如肿瘤坏死因子α和白细胞介素6)启动子通过特定的甲基转移酶SET和含有MYND结构域2(Smyd2)具有组蛋白3赖氨酸4和H3K36二甲基化作用,这可导致巨噬细胞M1极化核转录因子κB 和ERK信号传导降低[89]。在人单核细胞白血病细胞(THP-1)的低强度激光治疗(LLLT)中,肿瘤坏死因子α基因位点H3水平升高的组蛋白修饰同样伴随着M1极化和M1相关趋化因子和细胞因子的激活。总之,M1或M2极化所需基因的组蛋白修饰可能在各种疾病中具有治疗潜力[84]。在临床上研究者们可以开发某些抑制或者激活蛋白以及酶的药物来控制治疗炎症性疾病。 此外,有研究发现,补体C3a及其受体C3aR组成的C3a-C3aR轴能够增加牙周炎症组织中M1型巨噬细胞的数量及M1型巨噬细胞标志物的表达,并且可能通过调控巨噬细胞的极化状态介导牙周炎症和软组织破坏[90]。使用肿瘤坏死因子α对牙龈组织来源的间充质干细胞进行预处理发现,其可以增加外泌体上CD73的表达进而诱导M2巨噬细胞极化,同时外泌体 miR-1260b 可通过Wnt5a介导的核转录因子κB受体活化因子配体途径抑制破骨细胞生成,从而有助于炎症的消退并防止牙周组织中的骨质流失[91]。还有研究发现,双加氧酶TET1敲低通过下调核转录因子κB 信号通路的活性,减弱脂多糖/干扰素γ诱导人单核细胞白血病细胞中的M1巨噬细胞极化,为研究巨噬细胞极化调节牙周炎提供思路[92]。"
| [1] CHAPPLE CC, SRIVASTAVA M, HUNTER N. Failure of macrophage activation in destructive periodontal disease. J Pathol. 1998;186(3): 281-286. [2] HIENZ SA, PALIWAL S, IVANOVSKI S. Mechanisms of bone resorption in periodontitis. J Immunol Res. 2015;2015:615486. [3] MOSSER DM, EDWARDS JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958-969. [4] 周琦,孙慧娟,于栋华,等.巨噬细胞M1/M2型极化在不同疾病中的作用机制[J].中国药理学通报,2020,36(11):1502-1506. [5] MUNOZ J, AKHAVAN NS, MULLINS AP, et al. Macrophage polarization and osteoporosis: a review. Nutrients. 2020;12(10):2999. [6] ARANGO DUQUE G, DESCOTEAUX A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. [7] WATANABE S, ALEXANDER M, MISHARIN AV, et al. The role of macrophages in the resolution of inflammation. J Clin Invest. 2019; 129(7):2619-2628. [8] MURRAY PJ. Macrophage polarization. Annu Rev Physiol. 2017;79:541-566. [9] SHAPOURI-MOGHADDAM A, MOHAMMADIAN S, VAZINI H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425-6440. [10] FUNES SC, RIOS M, ESCOBAR-VERA J, et al. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186-195. [11] XU W, ZHAO X, DAHA MR, et al. Reversible differentiation of pro- and anti-inflammatory macrophages. Mol Immunol. 2013;53(3):179-186. [12] YAO Y, XU XH, JIN L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol. 2019;10:792. [13] 王乐旬,张盛昔,吴惠娟,等.巨噬细胞极化中特异性分子表达的实验研究[J].广东药科大学学报,2020,36(1):43-49. [14] COLIN S, CHINETTI-GBAGUIDI G, STAELS B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1):153-166. [15] KONG X, GAO J. Macrophage polarization: a key event in the secondary phase of acute spinal cord injury. J Cell Mol Med. 2017;21(5):941-954. [16] YASUDA H, SHIMA N, NAKAGAWA N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597-3602. [17] SIMA C, GLOGAUER M. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. Periodontol 2000. 2013;63(1):80-101. [18] ADAMOPOULOS IE, MELLINS ED. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11(3):189-194. [19] VAN RAEMDONCK K, UMAR S, PALASIEWICZ K, et al. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell Mol Life Sci. 2020;77(7):1387-1399. [20] MANTOVANI A, SICA A, SOZZANI S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677-686. [21] SUN X, GAOJ, MENG X, et al. Polarized macrophages in periodontitis: characteristics, function, and molecular signaling. Front Immunol. 2021;12:763334. [22] YU T, ZHAO L, HUANG X, et al. Enhanced activity of the macrophage m1/m2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol. 2016;87(9):1092-1102. [23] CROTTI TN, DHARMAPATNI AA, ALIAS E, et al. Osteoimmunology: major and costimulatory pathway expression associated with chronic inflammatory induced bone loss. J Immunol Res. 2015;2015:281287. [24] CHANG LY, LAI CH, KUO CH, et al. Recombinant thrombomodulin lectin-like domain attenuates Porphyromonas gingivalis lipopolysaccharide-induced osteoclastogenesis and periodontal bone resorption. J Periodontol. 2021;92(11):1622-1634. [25] HAJISHENGALLIS G, DARVEAU RP, CURTIS MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717-725. [26] BURNS E, BACHRACH G, SHAPIRA L, et al. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177(12):8296-8300. [27] PAGE RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontal Res. 1991;26(3 Pt 2):230-242. [28] 潘佳慧,唐秋玲,李格格,等.巨噬细胞极化在牙龈卟啉单胞菌促进牙周炎发生发展中的作用[J].国际口腔医学杂志,2017,44(5): 533-537. [29] LAM RS, O’BRIEN-SIMPSON NM, LENZO JC, et al. Macrophage depletion abates Porphyromonas gingivalis-induced alveolar bone resorption in mice. J Immunol. 2014;193(5):2349-2362. [30] ZHUANG Z, YOSHIZAWA-SMITH S, GLOWACKI A, et al. Induction of M2 macrophages prevents bone loss in murine periodontitis models. J Dent Res. 2019;98(2):200-208. [31] HE D, KOU X, LUO Q, et al. Enhanced M1/M2 macrophage ratio promotes orthodontic root resorption. J Dent Res. 2015;94(1):129-139. [32] 白林,辛月娇,段丁瑜,等.巨噬细胞功能和炎症消退机制及与牙周炎关系研究进展[J].华西口腔医学杂志,2017,35(4):427-432. [33] BANERJEE S, BIEHL A, GADINA M, et al. JAK-STAT Signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521-546. [34] 唐俭,陈旭昕,韩志海.巨噬细胞极化及极化调控的研究进展[J].转化医学杂志,2019,8(6):373-376. [35] SOUZA JA, ROSSA C Jr, GARLET GP, et al. Modulation of host cell signaling pathways as a therapeutic approach in periodontal disease. J Appl Oral Sci. 2012;20(2):128-138. [36] OH H, PARK SH, KANG MK, et al. Asaronic acid attenuates macrophage activation toward M1 phenotype through inhibition of NF-κB pathway and JAK-STAT signaling in glucose-loaded murine macrophages. J Agric Food Chem. 2019;67(36):10069-10078. [37] NI Y, ZHUGE F, NAGASHIMADA M, et al. Novel action of carotenoids on non-alcoholic fatty liver disease: macrophage polarization and liver homeostasis. Nutrients. 2016;8(7):391. [38] GARCIA DE AQUINO S, MANZOLLI LEITE FR, STACH-MACHADO DR, et al. Signaling pathways associated with the expression of inflammatory mediators activated during the course of two models of experimental periodontitis. Life Sci. 2009;84(21-22):745-754. [39] CHACES DE SOUZA JA, NOGUEIRA AV, CHAVES DE SOUZA PP, et al. SOCS3 expression correlates with severity of inflammation, expression of proinflammatory cytokines, and activation of STAT3 and p38 MAPK in LPS-induced inflammation in vivo. Mediators Inflamm. 2013;2013: 650812. [40] 张洁,田艾.M2巨噬细胞参与骨再生相关信号通路的作用与机制[J].中国组织工程研究,2023,27(2):314-321. [41] WANG N, GAO J, JIA M, et al. Exendin-4 induces bone marrow stromal cells migration through bone marrow-derived macrophages polarization via PKA-STAT3 signaling pathway. Cell Physiol Biochem. 2017;44(5):1696-1714. [42] 吴燕,张定然,王新慧,等.巨噬细胞极化及其对炎性疾病作用的研究进展[J].中国畜牧杂志,2021,57(7):22-26. [43] PAPADOPOULOS G, WEINBERG EO, MASSARI P, et al. Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J Immunol. 2013;190(3):1148-1157. [44] JAIN S, COATS SR, CHANG AM, et al. A novel class of lipoprotein lipase-sensitive molecules mediates Toll-like receptor 2 activation by Porphyromonas gingivalis. Infect Immun. 2013;81(4):1277-1286. [45] LAWRENCE T, NATOLI G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011; 11(11):750-761. [46] LINTON MF, MOSLEHI JJ, BABAEV VR. Akt signaling in macrophage polarization,survival,and atherosclerosis. Int J Mol Sci. 2019;20(11): 2703. [47] TROUTMAN TD, BAZAN JF, PASARE C. Toll-like receptors,signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle. 2012;11(19):3559-3553567. [48] ARRANZ A, DOXAKI C, VERGADI E, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A. 2012;109(24):9517-9522. [49] BAO L, LI X. MicroRNA-32 targeting PTEN enhances M2 macrophage polarization in the glioma microenvironment and further promotes the progression of glioma. Mol Cell Biochem. 2019;460(1-2):67-79. [50] VERGADI E, IERONYMAKI E, LYRONI K, et al. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol. 2017; 198(3):1006-1014. [51] WU L, GUO Q, Yang J, et al. Tumor necrosis factor alpha promotes osteoclast formation via PI3K/Akt pathway-mediated blimp1 expression upregulation. J Cell Biochem. 2017;118(6):1308-1315. [52] ZHAO SJ, KONG FQ, JIE J, et al. Macrophage MSR1 promotes BMSC osteogenic differentiation and M2-like polarization by activating PI3K/AKT/GSK3β/β-catenin pathway. Theranostics. 2020;10(1):17-35. [53] DISKIN C, PALSSON-MCDERMOTT EM. Metabolic modulation in macrophage effector function. Front Immunol. 2018;9:270. [54] MEISTER M, TOMASOVIC A, BANNING A, et al. Mitogen-activated protein (map) kinase scaffolding proteins: a recount. Int J Mol Sci. 2013; 14(3):4854-4884. [55] 赵娜,方慧,唐亚平,等.MAPK信号转导通路与慢性牙周炎的相关研究进展[J].口腔医学研究,2017,33(9):1012-1015. [56] LI Q, VALERIO MS, KIRKWOOD KL. MAPK usage in periodontal disease progression. J Signal Transduct. 2012;2012:308943. [57] WANG L, ZHENG J, PATHAK JL, et al. SLIT2 overexpression in periodontitis intensifies inflammation and alveolar bone loss, possibly via the activation of MAPK pathway. Front Cell Dev Biol. 2020;8:593. [58] SON HJ, EO HJ, PARK GH, et al. Heracleum moellendorffii root extracts exert immunostimulatory activity through TLR2/4-dependent MAPK activation in mouse macrophages, RAW264.7 cells. Food Sci Nutr. 2020;9(1):514-521. [59] SUN W, ZHANG H, WANG H, et al. Targeting notch-activated M1 macrophages attenuates joint tissue damage in a mouse model of inflammatory arthritis. J Bone Miner Res. 2017;32(7):1469-1480. [60] 刘利萍,张焱皓,李茂,等.调控巨噬细胞极化的相关信号通路及其调节机制研究进展[J].中国免疫学杂志,2021,37(6):747-753. [61] REGAN J, LONG F. Notch signaling and bone remodeling. Curr Osteoporos Rep. 2013;11(2):126-129. [62] KRAUSGRUBER T, BLAZEK K, SMALLIE T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231-238. [63] ZHAO B, TAKAMI M, YAMADA A, et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med. 2009;15(9):1066-1071. [64] ZHAO B, GRIMES SN, LI S, et al. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp Med. 2012;209(2):319-334. [65] SHAIK-DASTHAGIRISAHEB YB, HUANG N, GIBSON FC 3RD. Inflammatory response to Porphyromonas gingivalis partially requires interferon regulatory factor (IRF) 3. Innate Immun. 2014;20(3):312-319. [66] 魏文燕,陈建英.非编码RNA调控巨噬细胞极化的研究进展[J].山东医药,2018,58(29):90-94. [67] SELF-FORDHAM JB, NAQVI AR, UTTAMANI JR, et al. Microrna: dynamic regulators of macrophage polarization and plasticity. Front Immunol. 2017;8:1062. [68] LI J, JIN F, CAI M, et al. LncRNA nron inhibits bone resorption in periodontitis. J Dent Res. 2022;101(2):187-195. [69] WANG Q, XIE Y, HE Q, et al. LncRNA-Cox2 regulates macrophage polarization and inflammatory response through the CREB-C/EBPβ signaling pathway in septic mice. Int Immunopharmacol. 2021;101(Pt B): 108347. [70] MARQUES-ROCHA JL, SAMBLAS M, MILAGRO FI, et al. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29(9): 3595-3611. [71] DU W, WANG L, LIAO Z, et al. Circ_0085289 alleviates the progression of periodontitis by regulating let-7f-5p/SOCS6 pathway. Inflammation. 2021;44(4):1607-1619. [72] BANERJEE S, XIE N, CUI H, et al. MicroRNA let-7c regulates macrophage polarization. J Immunol. 2013;190(12):6542-6549. [73] ZHANG D, CAO X, LI J, et al. MiR-210 inhibits NF-κB signaling pathway by targeting DR6 in osteoarthritis.Sci Rep. 2015;5:12775. [74] MA S, LIU M, XU Z, et al. A double feedback loop mediated by microRNA-23a/27a/24-2 regulates M1 versus M2 macrophage polarization and thus regulates cancer progression. Oncotarget. 2016; 7(12):13502-13519. [75] GANTA VC, CHOI MH, KUTATELADZE A, et al. A microRNA93-interferon regulatory factor-9-immunoresponsive gene-1-itaconic acid pathway modulates M2-like macrophage polarization to revascularize ischemic muscle. Circulation. 2017;135(24):2403-2425. [76] SAHU SK, KUMAR M, CHAKRABORTY S, et al. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPβ regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog. 2017;13(5): e1006410. [77] JIANG Y, FU J, DU J, et al. DNA methylation alterations and their potential influence on macrophage in periodontitis. Oral Dis. 2022; 28(2):249-263. [78] JONES PA, LIANG G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10(11):805-811. [79] LINDROTH AM, PARK YJ. Epigenetic biomarkers: a step forward for understanding periodontitis. J Periodontal Implant Sci. 2013;43(3): 111-120. [80] LARSSON L, CASTILHO RM, GIANNOBILE WV. Epigenetics and its role in periodontal diseases: a state-of-the-art review. J Periodontol. 2015; 86(4):556-568. [81] YANG X, WANG X, LIU D, et al. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014;28(4): 565-574. [82] MIAO D, GODOVIKOVA V, QIAN X, et al. Treponema denticola upregulates MMP-2 activation in periodontal ligament cells: interplay between epigenetics and periodontal infection. Arch Oral Biol. 2014; 59(10):1056-1064. [83] WANG X, CAO Q, YU L, et al. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight. 2016;1(19):e87748. [84] ZHOU D, YANG K, CHEN L, et al. Promising landscape for regulating macrophage polarization: epigenetic viewpoint. Oncotarget. 2017; 8(34):57693-57706. [85] UEHARA O, ABIKO Y, SAITOH M, et al. Lipopolysaccharide extracted from Porphyromonas gingivalis induces DNA hypermethylation of runt-related transcription factor 2 in human periodontal fibroblasts. J Microbiol Immunol Infect. 2014;47(3):176-181. [86] DASKALAKI MG, TSATSANIS C, KAMPRANIS SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J Cell Physiol. 2018;233(9):6495-6507. [87] XUAN D, HAN Q, TU Q, et al. Epigenetic modulation in periodontitis: interaction of adiponectin and JMJD3-IRF4 axis in macrophages. J Cell Physiol. 2016;231(5):1090-1096. [88] GARRETT S, DIETZMANN-MAURER K, SONG L, et al. Polarization of primary human monocytes by IFN-gamma induces chromatin changes and recruits RNA Pol II to the TNF-alpha promoter. J Immunol. 2008;180(8):5257-5266. [89] XU G, LIU G, XIONG S, et al. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) production. J Biol Chem. 2015;290(9):5414-5423. [90] 任飞龙,罗环宇,郑适泽,等.C3a-C3aR轴通过巨噬细胞向M1型极化对慢性牙周炎模型小鼠炎症反应和组织损伤的影响[J].吉林大学学报(医学版),2022,48(1):1-8. [91] NAKAO Y, FUKUDA T, ZHANG Q, et al. Exosomes from TNF-α-treated human gingiva-derived MSCs enhance M2 macrophage polarization and inhibit periodontal bone loss. Acta Biomater. 2021;122:306-324. [92] HUANG Y, TIAN C, LI Q, et al. TET1 Knockdown Inhibits Porphyromonas gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization through the NF-κB Pathway in THP-1 Cells. Int J Mol Sci. 2019;20(8):2023. [93] RAMIREZ-CARROZZI VR, NAZARIAN AA, LI CC, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20(3):282-296. [94] BAKER RG, HAYDEN MS, GHOSH S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11-22. [95] XU H, ZHU J, SMITH S, et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat Immunol. 2012;13(7):642-650. [96] ZHONG Y, YI C. MicroRNA-720 suppresses M2 macrophage polarization by targeting GATA3. Biosci Rep. 2016;36(4):e00363. [97] LIANG YB, TANG H, CHEN ZB, et al. Downregulated SOCS1 expression activates the JAK1/STAT1 pathway and promotes polarization of macrophages into M1 type. Mol Med Rep. 2017;16(5):6405-6411. |
| [1] | Pan Zhongjie, Qin Zhihong, Zheng Tiejun, Ding Xiaofei, Liao Shijie. Targeting of non-coding RNAs in the pathogenesis of the osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1441-1447. |
| [2] | Dang Yi, Du Chengyan, Yao Honglin, Yuan Nenghua, Cao Jin, Xiong Shan, Zhang Dingmei, Wang Xin. Hormonal osteonecrosis and oxidative stress [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1469-1476. |
| [3] | Li Mengfei, Zhang Hong, Zhao Shaojian, Yin Guanghao, Wang Qibao. Expression of forkhead box protein 3 in refractory periapical periodontitis in rats with Enterococcus faecalis infection [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1187-1192. |
| [4] | Yang Zhishan, Tang Zhenglong. YAP/TAZ, a core factor of the Hippo signaling pathway, is involved in bone formation [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1264-1271. |
| [5] | Nie Chenchen, Su Kaiqi, Gao Jing, Fan Yongfu, Ruan Xiaodi, Yuan Jie, Duan Zhaoyuan, Feng Xiaodong. The regulatory role of circular RNAs in cerebral ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1286-1291. |
| [6] | Gao Yu, Han Jiahui, Ge Xin. Immunoinflammatory microenvironment after spinal cord ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1300-1305. |
| [7] | Yang Yitian, Wang Lu, Yao Wei, Zhao Bin. Application of the interaction between biological scaffolds and macrophages in bone regeneration [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1071-1079. |
| [8] | Li Wenjie, You Aijia, Zhou Junli, Fang Sujuan, Li Chun. Effects of different dressings in the treatment of burn wounds: a network meta-analysis [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1141-1148. |
| [9] | Yin Shuoxin, Zhang Tao, Wang Shuping, Lu Xin, Huang Xuping, Yin Mengying, Yang Yuwei, Yuan Bingmao, Mao Zhihua, Chen Yuanneng. Bibliometric and visualized analysis of researches on gastric cancer stem cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(6): 941-947. |
| [10] | Liu Wentao, Feng Xingchao, Yang Yi, Bai Shengbin. Effect of M2 macrophage-derived exosomes on osteogenic differentiation of bone marrow mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(6): 840-845. |
| [11] | Li Zhichao, Tan Guoqing, Su Hui, Xu Zhanwang, Xue Haipeng. Regulatory role of non-coding RNAs as potential therapeutic targets in spinal cord injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(5): 758-764. |
| [12] | Shao Zichen, Li Huanan, Gu Bing, Zhang Xiaoyun, Sun Weikang, Liu Yongqian, Gan Bin. MicroRNA, long non-coding RNA and circular RNA mediate the mechanism of decreasing uric acid, anti-inflammation and regulating bone metabolism in gout [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(5): 765-771. |
| [13] | Hu Xinming, Qiao Yanhua, Wang Xiaofan, Li Linyu, Zhao Bing. Mechanism of long non-coding RNA plasmacytoma variant translocation 1 involved in pelvic organ prolapse [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(5): 669-675. |
| [14] | Zhang Lichen, Chen Liang, Gu Yong. Inorganic ion bionic periosteum regulates immune microenvironment to promote bone repair [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(3): 346-353. |
| [15] | Dai Xianglin, Zhang Wenfeng, Yao Xijun, Shang Jiaqi, Huang Qiujin, Ren Yifan, Deng Jiupeng. Barium titanate/polylactic acid piezoelectric composite film affects adhesion, proliferation, and osteogenic differentiation of MC3T3-E1 cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(3): 367-373. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||