中国组织工程研究 ›› 2026, Vol. 30 ›› Issue (11): 2933-2948.doi: 10.12307/2026.305
• 组织构建相关数据分析 Date analysis of organization construction • 上一篇
糖尿病与肥厚型心肌病的因果关系:GWAS数据库信息分析
赵迎新1,2,郎 彤3,孟令丙4,5,6,高玉霞7
- 1天津医科大学研究生院,天津市 300203;2河北大学附属医院心内科,河北省保定市 071030;3潍坊市第二人民医院呼吸内科,山东省潍坊市 261041;4心脏代谢医学中心,国家心血管疾病临床医学研究中心,阜外医院,中国医学科学院及北京协和医学院,北京市 100037;5阜外医院心内科,国家心血管疾病临床医学研究中心,中国医学科学院及北京协和医学院,北京市 100037;6心血管疾病国家重点实验室,北京市 100037;7天津医科大学总医院心内科,天津市 300070
Causal relationship between diabetes mellitus and hypertrophic cardiomyopathy: information analysis based on the GWAS database
Zhao Yingxin1, 2, Lang Tong3, Meng Lingbing4, 5, 6, Gao Yuxia7
- 1Graduate School, Tianjin Medical University, Tianjin 300203, China; 2Department of Cardiology, Affiliated Hospital of Hebei University, Baoding 071030, Hebei Province, China; 3Department of Pneumology, Weifang Second People’s Hospital, Weifang 261041, Shandong Province, China; 4Cardiometabolic Medicine Center, National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital, National Clinical Research Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China; 5Department of Cardiology, Fuwai Hospital, National Clinical Research Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China; 6State Key Laboratory of Cardiovascular Disease, Beijing 100037, China; 7Department of Cardiology, Tianjin Medical University General Hospital, Tianjin 300070, China
摘要:

文题释义:
孟德尔随机化:是一种基于遗传变异的因果推断方法,在流行病学研究中广泛用于探索暴露因素与疾病结局之间的因果关系。孟德尔随机化通过遗传工具变量规避了传统观察性研究的混杂因素,提高了因果推断的可靠性。
HSF1:是一种关键的转录因子,调控细胞应激反应,主要参与热休克蛋白的表达调控。
背景:糖尿病与肥厚型心肌病相互影响,糖尿病患者心血管并发症风险显著升高,但两者之间的因果关系及分子机制尚不明确。
目的:利用孟德尔随机化方法,结合GWAS和GEO数据库数据,评估糖尿病对肥厚型心肌病的因果效应,通过生物信息学分析筛选关键调控因子。
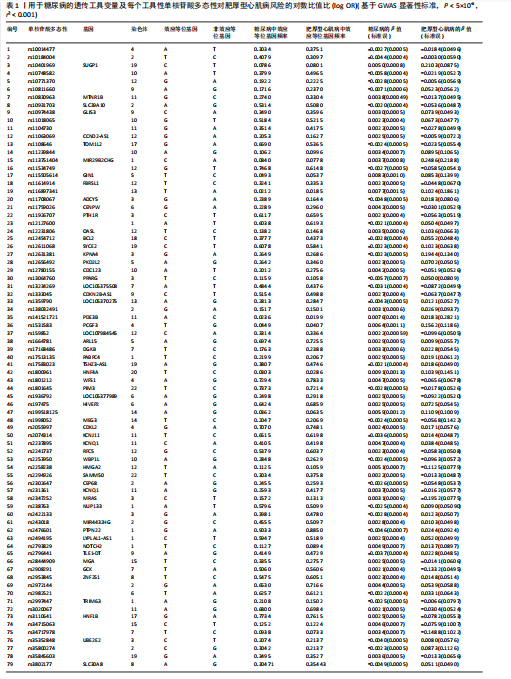
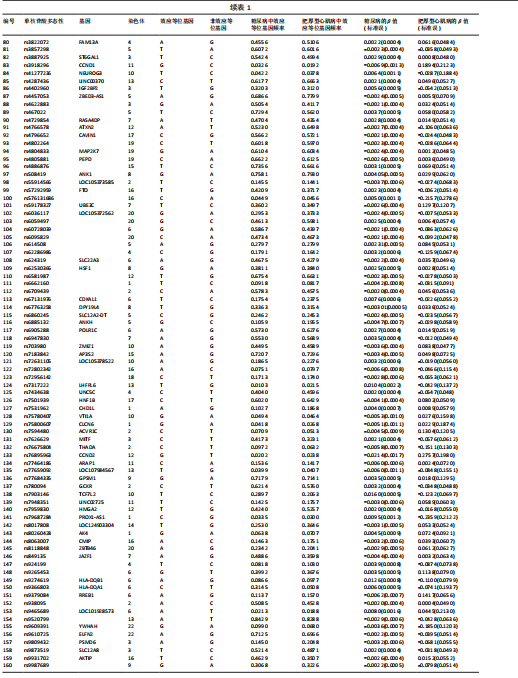
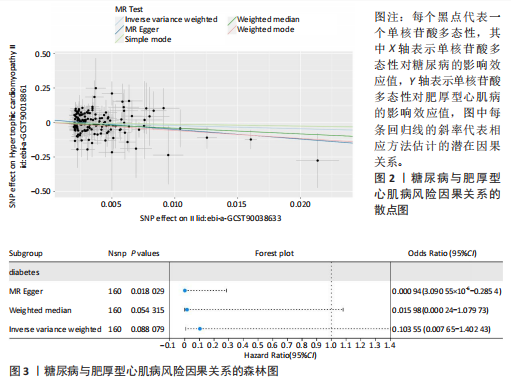
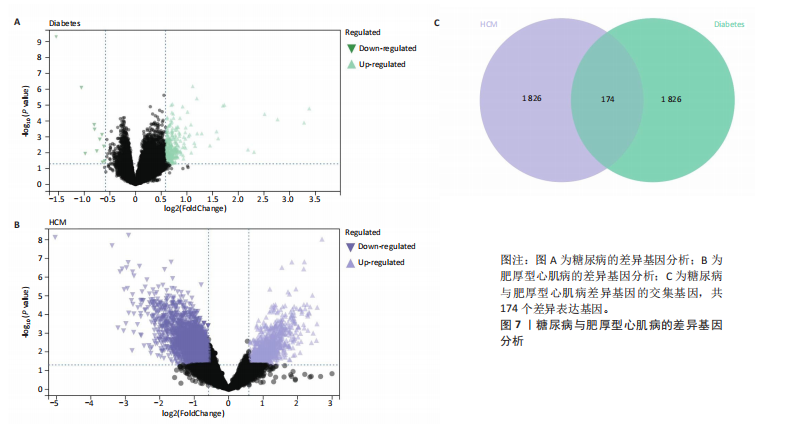
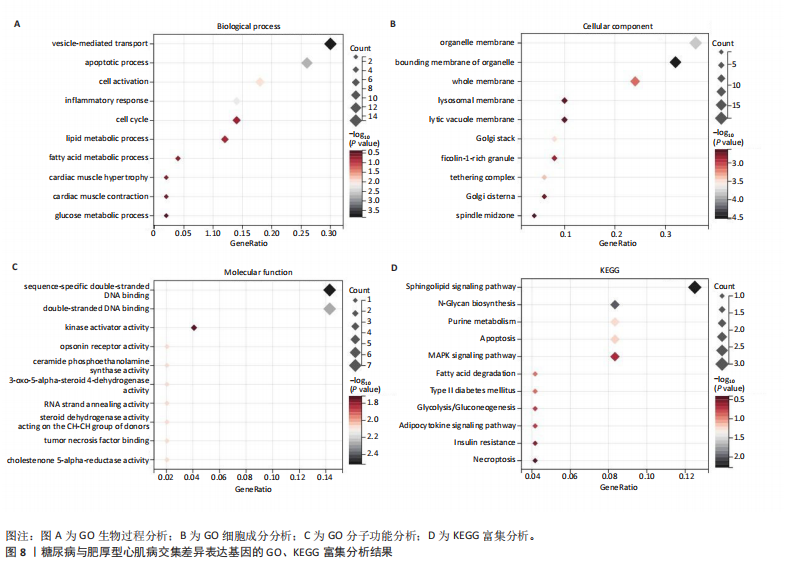
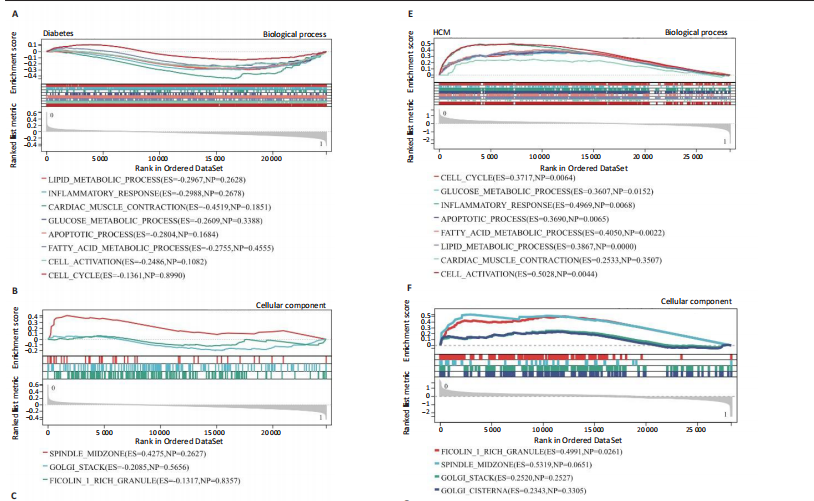
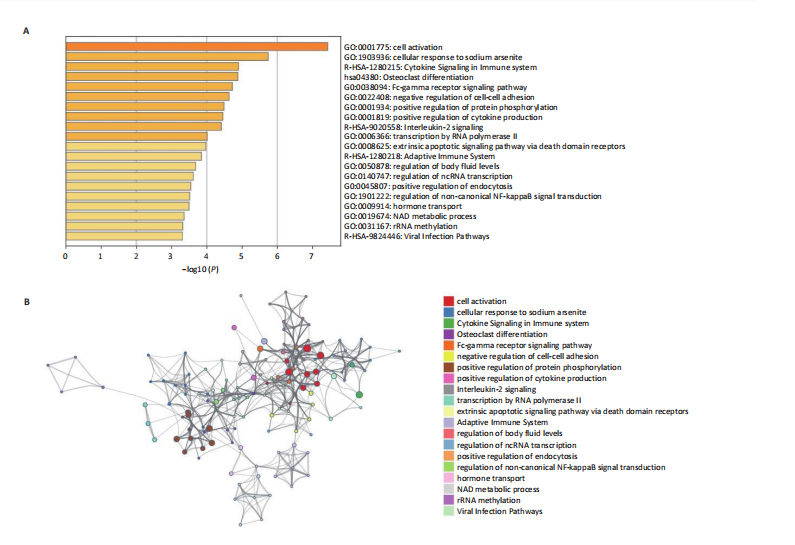
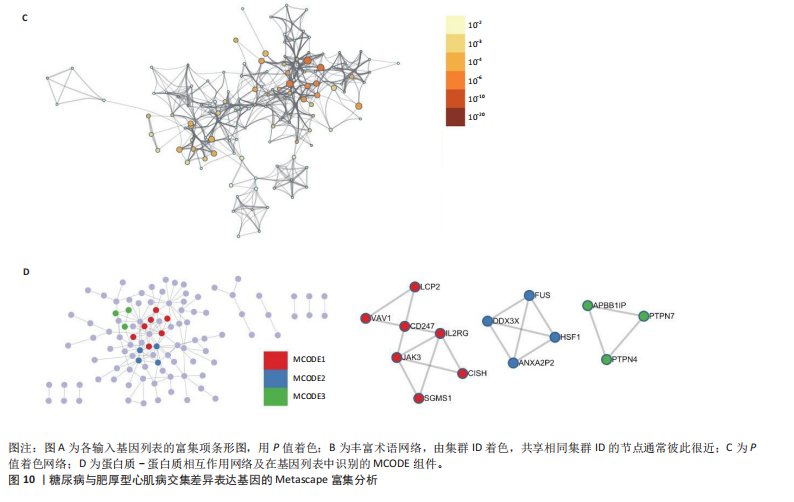
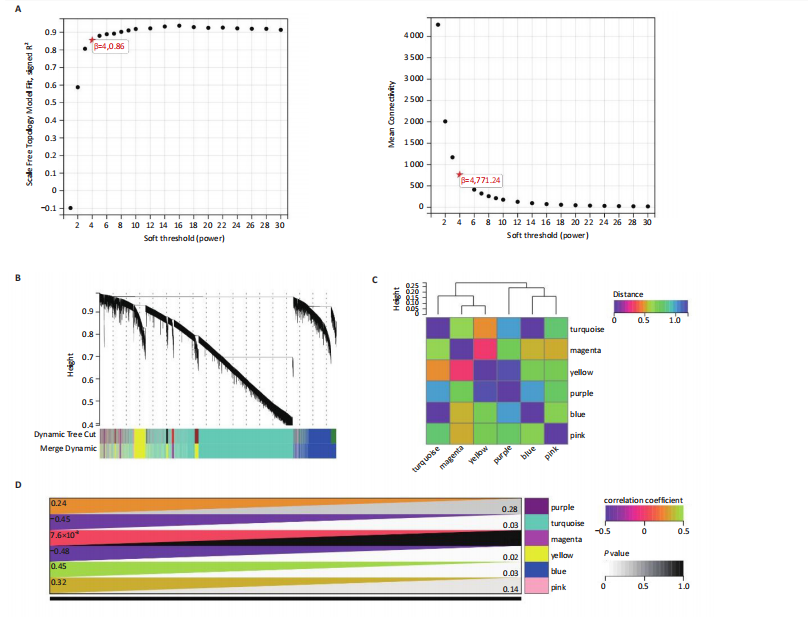
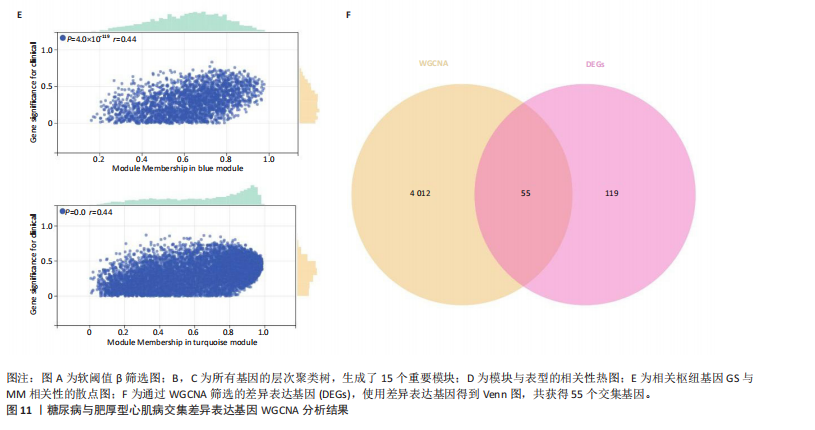
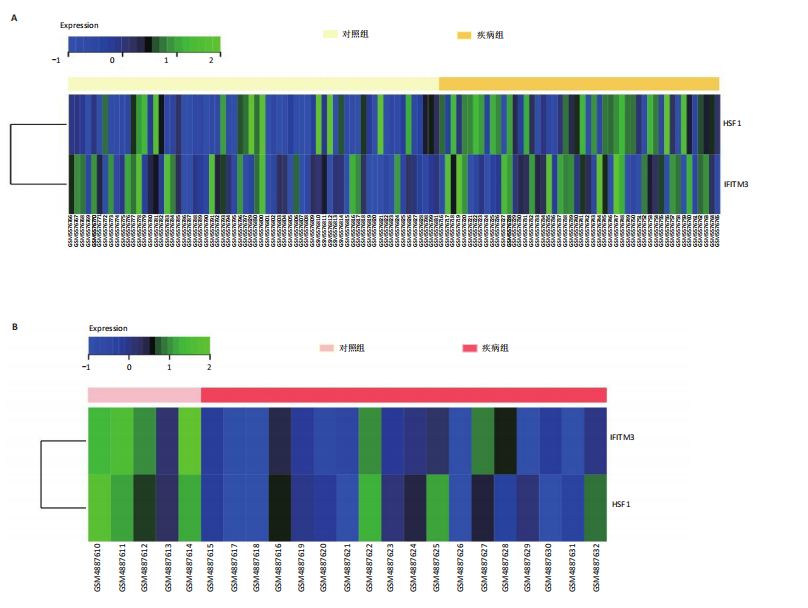
方法:从最新的全基因组关联研究(GWAS,由美国国家人类基因组研究所和欧洲生物信息研究所合作建立)数据库中获取162个与糖尿病相关的单核苷酸多态性,这些数据来自24 659例糖尿病患者和459 939名对照参与者,获取肥厚型心肌病数据,包括507例肥厚型心肌病病例和489 220名对照参与者,进行两样本孟德尔随机化分析,使用逆方差加权法评估糖尿病与肥厚型心肌病之间的因果关系。下载基因表达数据库(GEO)中的糖尿病数据集GSE184050和肥厚型心肌病数据集GSE160997,利用WGCNA探讨与糖尿病和肥厚型心肌病相关的重要模块及核心基因。通过GSEA探讨核心基因相关的富集术语和通路。将核心基因输入比较毒理学基因组数据库(CTD)网站以确定与核心基因相关的疾病。采用多种算法探讨HSF1在糖尿病和肥厚型心肌病中的作用。
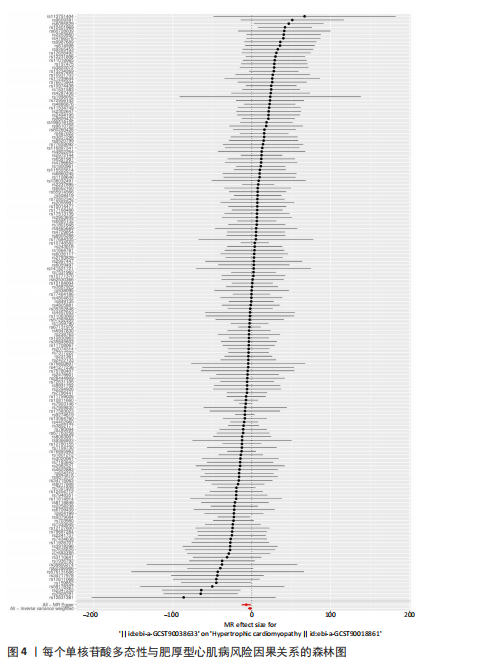
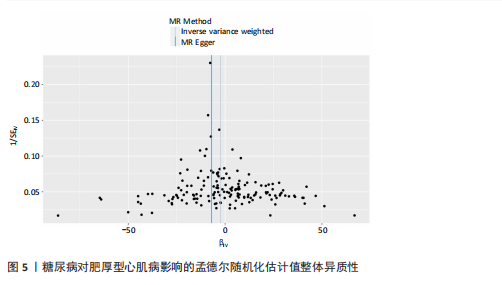
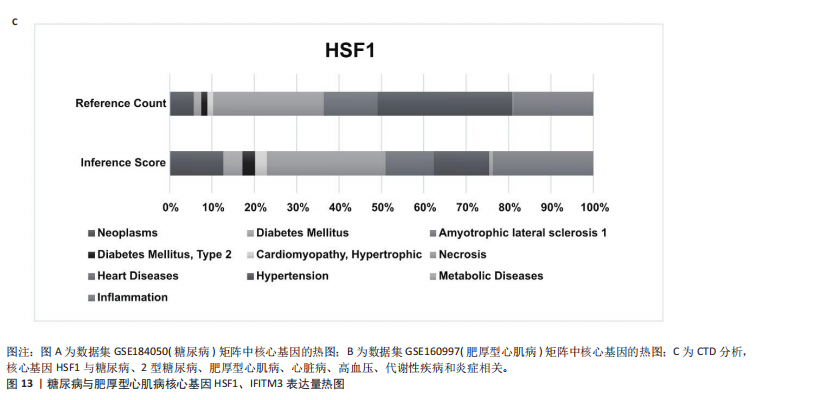
结果与结论:①以糖尿病为暴露因子,提取了160个单核苷酸多态性的分析结果,采用逆方差加权、MR-Egger和加权中位数回归方法评估遗传预测的糖尿病与肥厚型心肌病之间的因果关系,MR-Egger检验显示糖尿病与肥厚型心肌病之间存在显著因果关联,确定HSF1为糖尿病与肥厚型心肌病因果关系的核心生物标志物(P=0.018 028 98),加权中位数和逆方差加权法也提供了一致趋势。通过GEO数据库的数据验证确认了HSF1是同时影响糖尿病和肥厚型心肌病的潜在核心生物标志物。HSF1在糖尿病中高表达、在肥厚型心肌病中低表达。通过逐一移除单核苷酸多态性并重复孟德尔随机化分析的敏感性分析显示,没有单一单核苷酸多态性显著影响因果关系估计。将HSF1输入CTD网站以识别与核心基因相关的疾病,结果显示HSF1与糖尿病、2型糖尿病、肥厚型心肌病、心脏病、高血压、代谢紊乱和炎症等相关联。②研究主要基于国际大型数据库(如欧洲人群的GWAS和GEO表达谱数据),采用遗传工具变量进行因果推断,具有较强的科学性。中国生物医学研究应加强本土多组学数据的积累和整合,借助类似方法探索适用于中国人群的精准预防和治疗靶点。
https://orcid.org/0000-0004-1761-2484 (赵迎新)
中国组织工程研究杂志出版内容重点:干细胞;骨髓干细胞;造血干细胞;脂肪干细胞;肿瘤干细胞;胚胎干细胞;脐带脐血干细胞;干细胞诱导;干细胞分化;组织工程
中图分类号: