Chinese Journal of Tissue Engineering Research ›› 2024, Vol. 28 ›› Issue (8): 1301-1306.doi: 10.12307/2024.228
Previous Articles Next Articles
Abnormal modification of alpha-synuclein and its mechanism in Parkinson’s disease
Qi Xue1, Li Jiahui1, Zhu Yuanfeng1, Yu Lu1, Wang Peng1, 2
- 1Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China; 2Laboratory of Neurodegenerative Diseases, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China
-
Received:2023-03-10Accepted:2023-03-29Online:2024-03-18Published:2023-07-19 -
Contact:Wang Peng, PhD, Associate professor, Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin132013, Jilin Province, China; Laboratory of Neurodegenerative Diseases, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China -
About author:Qi Xue, Master candidate, Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China -
Supported by:Natural Science Foundation of Jilin Province, No. YDZJ202201ZYTS575 (to WP); Jilin Province Health and Health Technology Innovation Project, No. 2018J083 (to WP); Postgraduate Innovative Project of Beihua University, No. 2021017 (to QX)
CLC Number:
Cite this article
Qi Xue, Li Jiahui, Zhu Yuanfeng, Yu Lu, Wang Peng. Abnormal modification of alpha-synuclein and its mechanism in Parkinson’s disease[J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1301-1306.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
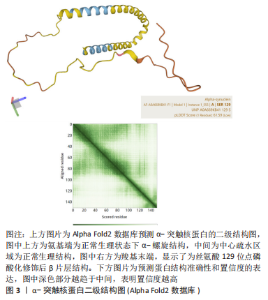
2.1 α-突触核蛋白的结构和功能 突触核蛋白主要位于神经突触和神经元核膜上,包括有有α,β,γ共3种类型,目前仅α-突触核蛋白被发现与帕金森病的发病密切相关[8]。在生理状态下α-突触核蛋白相对分子质量约为16 000的无序状蛋白单体,由140个氨基酸组成,分为氨基端、中心疏水区域和羧基末端3个功能区,见图3。在病理状态下3个功能区局部肽段发生突变、缺失和错误折叠等因素进而影响蛋白的结构特征,变成不可溶性聚集体。"


α-突触核蛋白的氨基端区域为第1-60氨基酸残基,由11个特征性的高度保守的残基组成7个重复序列(核心为KTKEGV),该重复序列可介导α-突触核蛋白以非内吞方式通过胞膜进入胞内[9]。氨基端主要带正电荷,由4个不完全载脂蛋白A1样重复序列组成,可介导脂质膜相结合形成类似的两亲性α-螺旋结构,因此泛素化和乙酰化的翻译后修饰多发生在此部位。在氨基端含有5个常见家族性帕金森病的突变位点,分别为A53T,A30P,E46K,G51D和H50Q。突变型中的3个(A30P,E46K和A53T)均可以促进 α-突触核蛋白寡聚体的形成,其他2个E46K和A53T可进一步加速促进聚集体的纤维化[10]。最近BELL等[11]通过生物物理方法测定氨基端乙酰化与脂质膜上α-突触核蛋白突变体之间联系,结果显示氨基端乙酰化修饰后5种突变体均促进合成淀粉样纤维结构,另外由于突变导致氨基端膜电荷发生显著变化,E46K在脂质囊泡中的聚集率显著高,与以往研究氨基端E46K突变导致错误折叠形成更稳定原纤维结构相一致[12]。MCGLINCHEY等[13]通过低温电子显微镜对α-突触核蛋白的结构进行检测观察,结果显示氨基端13,35和40残基的缺失可促进蛋白原纤维形态产生。因此,α-突触核蛋白氨基端异常修饰的发病机制对探索帕金森病发病起决定性因素。 α-突触核蛋白中心疏水区域为61-95氨基酸残基,具有疏水区域特性易形成β片层结构,可促进蛋白整体发生聚集。KAPASI等[14]通过全外显子组测序检测α-突触核蛋白中央的疏水区域发现一种新的突变体(E83Q)与路易体痴呆和阿尔茨海默综合征等突触核蛋白密切相关,随后对患者临床诊断评估,并使用组织病理学检测患者死后脑内额颞叶病理变化,结果显示患者表现出失语症临床特征和严重的非典型额颞叶萎缩。KUMAR等[15]使用生物化学和生物物理方法进行体外实验与细胞模型相结合,结果表明位于中央的疏水区域残基(61-95)的突变体促进原纤维接种活性,并促进类似帕金森患者脑内多种形态如环状的路易小体包涵体生成。此外NASSTROM等[16]利用分子动力学模拟设计α-突触核蛋白中央的疏水区域残基(71-84)合成肽段,以产生更多原纤维结构,结果证明可模拟该区域蛋白与帕金森疾病相关的病理形态特征如柱状和多晶型,该结论表明对蛋白特定区域的病理改变,研究者们可以借助数字技术直观观察并模拟蛋白构象改变。 α-突触核蛋白羧基末端为96-140位氨基酸,富含大量带负电荷的酸性氨基酸和脯氨酸,因易与金属离子相互作用而具有较强的亲水性,因此是主要的磷酸化修饰区域[17]。DASARI等[18]采用生物物理技术系统地研究了带有正电荷含磷酸基蛋白和α-突触核蛋白带负电荷的羧基端混合物的聚集过程,结果表明含磷酸基蛋白的单体形式选择性地与α-突触核蛋白的羧基端区域相互作用,导致α-突触核蛋白错误折叠形成具有不同分子构象的不同丝状聚集体。VAN DER WATEREN等[19]研究发现在帕金森患者脑内α-突触核蛋白羧基末端两个区域(1-119)和(1-103)截断后,由于截断部位pH值存在差异脂质,进而诱导蛋白聚集机制产生变化,具有生理pH值的区域会完成初级原纤维向成熟原纤维转化。 在正常生理情况下,α-突触核蛋白为无定型结构的小分子单体,呈无规则卷曲,不发生折叠[20],其单体结构并不稳定。α-突触核蛋白在生理条件发生变化时经历了多种翻译后修饰,这些修饰对细胞毒性作用和α-突触核蛋白形成聚集体的聚集率均有影响,在不同的环境条件下显示出较强的构象可塑性,主要表现为形成低聚物、原纤维和淀粉样纤维这3种结构[21]。因此在帕金森病病理条件下,α-突触核蛋白发生错误折叠,其结构会转变为 β 片层,并逐步形成小的可溶性寡聚体、大分子量的不溶的前体纤维,具有较强的神经毒性。可见α-突触核蛋白的功能区二级结构的差异使得不同位点修饰对整个蛋白构象重塑和进一步聚集的影响差异巨大。下面进一步讨论α-突触核蛋白发生不同翻译后修饰的机制,以及其产生的作用,见表1。"


2.2 α-突触核蛋白的磷酸化修饰 2.2.1 修饰位点对α-突触核蛋白的作用 α-突触核蛋白磷酸化修饰在翻译后修饰最常见。在磷酸化修饰中丝氨酸、苏氨酸和酪氨酸磷酸化较常见,其中丝氨酸磷酸化占75%以上。早在2015年,李蕾等[22]通过质谱分析可检测到的高度可信的磷酸化修饰位点共有2个,丝氨酸87和酪氨酸125。α-突触核蛋白在生理状态下氨基端为α-螺旋结构,C端因亲水性较强易磷酸化,从而形成β片层,促进蛋白易于聚集[23],见图3。 在α-突触核蛋白翻译后修饰中,残基丝氨酸129处的磷酸化及其对α-突触核蛋白病的影响具有重要的研究价值,并被认为在调节α-突触核蛋白聚集和神经毒性中起关键作用。有研究表明帕金森患者脑内约有90%的α-突触核蛋白发生丝氨酸129位点磷酸化修饰,正常大脑中仅占不到4%[24]。在2006年,ANDERSON等[25]通过酶联免疫吸附剂法和质谱检测确定丝氨酸129磷酸化是家族型和散发型路易体患者脑内α-突触核蛋白主要的翻译后修饰。并且路易小体内存在含量较高的丝氨酸129磷酸化的α-突触核蛋白,是构成淀粉样包涵体的基础[26]。 GABRIELYAN等[27]使用过表达的重组人α-突触核蛋白构建帕金森病小鼠模型,通过行为学检测和免疫组织化学方法观察,丝氨酸129磷酸化的α-突触核蛋白在雄性小鼠模型内中脑黑质致密部诱导的病理改变与人脑并无差异,进一步表明动物模型可有效表达疾病的病理改变,但α-突触核蛋白的特定构象与其在的大脑区域特异性分布还需进一步研究。 磷酸化修饰后的蛋白在体内体外对细胞表现出不一样的生物学作用。其中丝氨酸129位点磷酸化修饰后的α-突触核蛋白的致病相关性饱受争议。DING等[28]应用甲基苯丙胺进行造模,在体内和体外模型内加入丝氨酸129磷酸化修饰后的蛋白,可发生朊病毒样弥散、诱导α-突触核蛋白的聚集,增加其播散能力,从而降低神经元细胞活力。但另体外研究发现,聚集初的丝氨酸129磷酸化修饰,反而会降低α-突触核蛋白聚集倾向,并抑制原纤维的形成,减弱细胞毒性[29]。这意味着α-突触核蛋白丝氨酸129磷酸化可能参与神经元保护机制,但是帕金森是一种进行性疾病,随着病程进展,患者脑内磷酸化修饰持续升高,丝氨酸129仅在患者患病初期具有保护作用。 VUIDE等[30]采取机器分类技术检测孵育7 d的帕金森患者中脑多巴胺能神经元,结果证实神经元中存在的树突分支较少,而丝氨酸129磷酸化α-突触核蛋白的含量和修饰程度不断加重,修饰后的蛋白对细胞具有较强的毒性作用。对此,CARIULO等[31]使用超灵敏免疫测定法检测帕金森病患者血液内丝氨酸129磷酸化的α-突触核蛋白,结果显示其特异度较高,对蛋白质磷酸酶极其敏感。虽然在血液内可特异性识别磷酸化后的蛋白,但无法分辨其为病理性还是生理性蛋白,对此ARLINGHAUS等[32]使用新方法(邻近连接测定)特异性检测细胞培养、小鼠和人脑切片中生理性丝氨酸129磷酸化的α-突触核蛋白,为更好地了解和探索帕金森病提供一个新工具。 2.2.2 激酶对磷酸化修饰的作用 磷酸化代谢可加速神经退行性疾病的病程进展,α-突触核蛋白的磷酸化修饰还与激酶的催化作用相关。G蛋白偶联受体激酶、蛋白酪氨酸激酶(c-Fgr,Syk,Lyn,Fyn和Src)和蛋白磷酸激酶等均为与磷酸化修饰相关激酶。WU等[33]检测转基因小鼠脑组织中G蛋白偶联受体激酶6和酪蛋白激酶2受一氧化氮调控S-亚硝基化后,丝氨酸129磷酸化蛋白水平上调,促进聚集形成路易小体。除此之外,SANO等[34]发现,在患有路易体痴呆的大脑中,酪蛋白激酶2可介导α-突触核蛋白在酪氨酸136处磷酸化。polo样激酶是主要调节丝氨酸/苏氨酸修饰的激酶。在帕金森病模型中证明蛋白磷酸酶2A与葡萄糖脑苷脂酶呈正相关[35],有研究在小鼠动物模型内可调控腺苷5’-单磷酸活化蛋白激酶/糖原合酶激酶3/蛋白磷酸酶2A相关蛋白通路,来抑制帕金森病程的进展,而且表明帕金森病中神经元凋亡与线粒体障碍密切相关[36]。早有研究证实神经酰胺可天然特异性激活线粒体蛋白磷酸酶2A[37],它的激活与细胞凋亡密切相关。另外,已有相关研究证实,帕金森病患者的血清由于内环境的产生病理改变,促使血清中PLK2活性的升高[38],可促进α-突触核蛋白磷酸化修饰水平上调,产生神经毒性。磷酸化修饰后蛋白产生的具体作用仍存在较大争议,需进一步结合相关帕金森病临床表现与病理结果来探究。 2.3 α-突触核蛋白的泛素化修饰 2.3.1 激酶对泛素化修饰的作用 低分子量的泛素蛋白通过特定的酶(能量依赖性激活酶、结合酶和选择性连接酶)与靶蛋白进行特异性修饰,进而被26S蛋白酶体识别、降解的过程称为泛素化 [39]。α-突触核蛋白含有15个赖氨酸残基,其中4个赖氨酸21、赖氨酸23、赖氨酸32和赖氨酸34位于重复序列的氨基末端,在体外易于泛素化,影响蛋白的膜穿梭能力。α-突触核蛋白的赖氨酸6、赖氨酸10和赖氨酸12位于氨基末端区域在体内易于发生泛素化。MOON等[40]检测在体外泛素化修饰后诱导α-突触核蛋白聚集体间的结构差异,研究发现在赖氨酸6、赖氨酸23和赖氨酸96位点的泛素化修饰会导致α-突触核蛋白的构象发生变化,均抑制α-突触核蛋白的聚集。此前MEIER等[41]的研究中也阐明了通过蛋白质半合成生成具有9个特异性泛素修饰的α-突触核蛋白衍生物,证明泛素化修饰位点的差异影响蛋白的聚集情况。正常机体内α-突触核蛋白几乎不发生泛素化,仅在路易体中可检测到,与磷酸化修饰程度正相关。因此,泛素化在帕金森病的病程中很可能发生在α-突触核蛋白磷酸化聚集后,参与路易小体的形成过程。在家族性帕金森病(α-突触核蛋白A53T突变)α-突触核蛋白中常同时发生磷酸化、特异性截断和泛素化修饰,且与散发性路易体病的发病机制有相似的途径[42]。王洪财[43]在鱼藤酮介导的帕金森病细胞和动物模型中发现泛素连接酶受神经营养素受体P75表达抑制后,调控α-突触核蛋白泛素化介导蛋白聚集,参与多巴胺能损伤调控,可能是P75干预治疗帕金森病的新靶点,但泛素化翻译后修饰的机制仍需研究者们进一步探索。 2.3.2 蛋白降解途径对泛素化修饰的作用 α-突触核蛋白胞内降解主要通过自噬-溶酶体途径和泛素-蛋白酶体系统这两个途径,泛素-蛋白酶体系统通过将小分子(泛素)附着到底物上来诱导蛋白酶体依赖性降解来影响蛋白质稳态,是α-突触核蛋白主要的降解途径。目前,体内和体外实验均证实家族性和散发性帕金森病可能与泛素蛋白酶体系统的功能缺陷有关,使α-突触核蛋白清除能力减弱,导致蛋白异常聚集,进而产生细胞毒性[44]。QUINN 等[45]研究发现parkin蛋白泛素化的减少会导致蛋白稳定性减弱进而连接酶失活,导致蛋白酶体再循环受损和自噬酶(程序性死亡受体1)相互作用的减少;而酪氨酸激酶抑制会增加parkin泛素化与自噬酶的作用,促进α-突触核蛋白自噬降解和黑质内神经元的存活。PARK等[46]也证明在泛素-蛋白酶体系统中去泛素化酶1可参与α-突触核蛋白调节,上调神经前体细胞发育下调蛋白1破坏α-突触核蛋白的结构稳定性,进一步提示其可能为帕金森病中α-突触核蛋白新的调控因子。 2.4 α-突触核蛋白的乙酰化修饰 2.4.1 蛋白结构对乙酰化修饰的影响 乙酰化是改变蛋白质功能最主要的修饰方式之一。α-突触核蛋白的氨基末端含大量赖氨酸和正电荷,是发生乙酰化的主要部位。α-突触核蛋白经乙酰化修饰,可以非折叠的、稳定可溶的α-螺旋四聚体(相对分子质量58 000-60 000)存在,是神经元内α-突触核蛋白的非变性聚集体形式[47]。病理状态下,赖氨酸侧链的乙酰化使N端正电荷减少,改变氨基端与带有负电荷的羧基端的黏附性,易于与羧基端发生排斥,增加中心疏水区的外显,提升膜穿梭能力,减少胞内的聚集。乙酰化的α-突触核蛋白在囊泡和突触膜中表现出强烈的脂质亲和力,改变膜结合特异性,增强α-突触核蛋白与神经节苷脂 1的结合,将原纤维形成速率减慢了约75%[48]。而且近期BELL等[49]采用动力学方法也观察到氨基末端发生乙酰化修饰会致使脂质诱导的聚集速率降低,并延缓原纤维伸长和聚集体增殖,并影响其聚集体的结构特性。翟紫凝等[50]认为,在蛋白的氨基端和羧基端都高度带负电荷,蛋白乙酰化修饰后,静电排斥占据主导作用,抑制蛋白纤维化聚集,也为蛋白乙酰化修饰提供一种新的抑制聚集的方法。但另一方面,部分位点乙酰化(14-31,50-57)导致静电力减弱,可降低对α-突触核蛋白单体或小低聚物的纠缠保护,促进α-突触核蛋白的纤维化[51-52]。 2.4.2 突变对乙酰化修饰的影响 家族突变型A53T -突触核蛋白氨基端乙酰化的位置有异于天然型,其氨基端残基1-12发生乙酰化修饰后易于与铁离子结合,可瞬时抑制螺旋形成,减少α-突触核蛋白原纤维形成;而在残基5-8,14-31和50-57时瞬时促进螺旋形成,增加原纤维的形成率[52]。除A53T突变外,TENG等[53]研究证实病理性H50Q突变不会提高铜离子与α-突触核蛋白的亲和力位点结合,反而减弱蛋白与铜离子的相互作用,与蛋白聚集,推翻富含铜离子的蛋白促进活性氧会导致损伤神经元该论点,为H50Q突变诱导帕金森病理改变提供新的见解。这些结果表明乙酰化修饰后的蛋白自身可能会减缓或抑制蛋白聚集,但乙酰化修饰后的蛋白与其他物质相结合,反而会再次二次成核,形成更稳定的原纤维结构。也同样表明翻译后修饰发生在不同位点,对蛋白聚集的影响差异显著,因而蛋白乙酰化修饰对帕金森病的发病机制仍需进一步探索。 2.5 α-突触核蛋白的硝基化修饰 2.5.1 修饰位点对α-突触核蛋白的作用 硝基化是蛋白质翻译后的不可或缺的修饰。该反应主要与信号分子(一氧化氮)作用后调节神经元的信号通路,在氧化剂的作用下转化成不同的活性氮物种,诱导蛋白质的硝基化,其主要修饰位点发生在酪氨酸和色氨酸[54]。酪氨酸在α-突触核蛋白囊泡结合中起重要作用,由于氨基端带负电荷,硝基化修饰后叠加负电荷,进而损害这种相互作用,打破蛋白内部动态平衡,从而促使蛋白低聚物产生。而且α-突触核蛋白与亚硝酸盐孵育后,可观察到4个酪氨酸残基(酪氨酸 39,酪氨酸 125,酪氨酸133,酪氨酸136)被硝基化修饰,酪氨酸125硝基化特异性靶点,可改变蛋白质的结构,有助于α-突触核蛋白二聚体和原纤维的形成。而酪氨酸39硝基化修饰提升α-突触核蛋白聚集效率,诱发神经毒性,损害多巴胺能神经元[55]。 2.5.2 氧化应激对硝基化修饰的影响 硝基化α-突触核蛋白的细胞毒性主要是通过整合素介导的诱导型一氧化氮合酶/局部粘着斑激酶途径来表达[56],将硝基化α-突触核蛋白加入人类神经母细胞瘤细胞一起培养时,可致使细胞死亡,而抗整合素 ɑ5β1可保护细胞。MUSGROVE等[57]使用百草枯建细胞和动物模型模拟氧化应激,结果证实氧化应激后影响硝基化修饰的蛋白在细胞内传递,促进蛋白向大脑其他区域扩散,证实了硝基化修饰后的蛋白在氧化应激下具有细胞间迁移能力,该机制可促使帕金森病的发生与发展。此外,STYKEL等[58]也实证当α-突触核蛋白的羧基端发生乙酰化修饰后会抑制线粒体与微管蛋白结合,阻断帕金森人类多能干细胞中的轴突线粒体转运。而且在前期研究中,GIASSON等[59]的研究中阐述了酪氨酸残基的硝基化可降低α-突触核蛋白的可溶性,促进α-突触核蛋白的原纤维形成,活性氧和氮形成的硝化剂可以氧化残留的酪氨酸残基并提高α-突触核蛋白聚合物的稳定性。该研究提供了硝基化和氧化应激诱导帕金森病发生的直接证据。SANTOS-LOBATO等[60]在帕金森诱导的运动障碍并发症患者的血浆和脑脊液中检测到一氧化氮的产物亚硝酸盐和硝酸盐的含量明显高于正常人。 硝基化α-突触核蛋白广泛存在与帕金森病患者的脑,胃肠道和血细胞内,但MA等[61]在患者的唾液内发现修饰后的蛋白,且患者病程进展均在早期阶段,这为帕金森的诊断提供一种新的标志物,而这些发现有助于进一步探究氧化应激对帕金森的作用。迄今为止,对帕金森病中发生的蛋白质(尤其是组蛋白)乙酰化修饰变化机制的研究非常有限。需要更系统和深入的研究来探索与帕金森病理学相关的特定乙酰化或去乙酰化及其潜在机制,见表2。"

| [1] GOEDERT M, JAKES R, SPILLANTINI MG. The Synucleinopathies: twenty years on. J Parkinsons Dis. 2017;7(s1):S51-S69. [2] WEINTRAUB D, MAMIKONYAN E. The neuropsychiatry of Parkinson disease: a perfect storm. Am J Geriatr Psychiatry. 2019;27(9):998-1018. [3] BLESA J, FOFFANI G, DEHAY B, et al. Motor and non-motor circuit disturbances in early Parkinson disease: which happens first? Nat Rev Neurosci. 2022;23(2):115-128. [4] POYMEROPOULOS MH, LAVEDAN C, LEROY E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045-2047. [5] WON SJ, FONG R, BUTLER N, et al. Neuronal oxidative stress promotes α-synuclein aggregation in vivo. Antioxidants (Basel). 2022;11(12):2466. [6] STEFANIS L, EMMANOILIDOU E, PANTAZOPOULOU M, et al. How is alpha‐synuclein cleared from the cell? J Neurochem. 2019;150:577-590. [7] YOO H, LEE J, KIM B, et al. Role of post-translational modifications on the alpha-synuclein aggregation-related pathogenesis of Parkinson’s disease. BMB Rep. 2022; 55(7):323-335. [8] HENDERSON MX, TROJANOWSKI JQ, LEE MY. α-synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci Lett. 2019;709:134316. [9] SARCHIONE A, MARCHAND A, TAYMANS JM, et al. Alpha-synuclein and lipids: the elephant in the room? Cells. 2021;10(9):2452. [10] ZHAO K, LI Y, LIU Z, et al. Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure. Nat Commun. 2020;11(1):2643. [11] BELL R, CASTELLANA-CRUZ M, NENE A, et al. Effects of n-terminal acetylation on the aggregation of disease-related α-synuclein variants. J Mol Biol. 2023;435(1):167825. [12] HOLEC SAM, LEE J, OEHLER A, et al. The E46K mutation modulates α-synuclein prion replication in transgenic mice. PLoS Pathog. 2022;18(12):e1010956. [13] MCGLINCHEY RP, NI X, SHADISH JA, et al. The N terminus of α-synuclein dictates fibril formation. Proc Natl Acad Sci U S A. 2021;118(35):e2023487118. [14] KAPASI A, BROSCH JR, NUDELMAN KN, et al. A novel SNCA E83Q mutation in a case of dementia with Lewy bodies and atypical frontotemporal lobar degeneration. Neuropathology. 2020;40(6):620-626. [15] KUMAR ST, MAHUL-MELLIER AL, HEGDE RN, et al. A NAC domain mutation (E83Q) unlocks the pathogenicity of human alpha-synuclein and recapitulates its pathological diversity. Sci Adv. 2022;8(17):eabn0044. [16] NASSTROM T, DAHLBERG T, MALYSHEV D, et al. Synthetic NAC 71-82 peptides designed to produce fibrils with different protofilament interface contacts. Int J Mol Sci. 2021;22(17):9334. [17] KACHAPPILLY N, SRIVASTAVA J, SWAIN BP, et al. Interaction of alpha-synuclein with lipids. Methods Cell Biol. 2022;169:43-66. [18] DASARI AKR, KAYED R, WI S, et al. Tau interacts with the C-terminal region of α-synuclein, promoting formation of toxic aggregates with distinct molecular conformations. Biochemistry. 2019;58(25):2814 -2821. [19] VAN DER WATEREN IM, KNOELES TPJ, BUELL AK, et al. C-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH. Chem Sci. 2018;9(25):5506-5516. [20] ESTAUN-PANZANO J, AROTCARENA ML, BEZARD E. Monitoring α-synuclein aggregation. Neurobiol Dis. 2023;176:105966. [21] 李冬青,秦晓红,米立志.α-突触核蛋白的结构生物学研究[J].中国生物化学与分子生物学报,2023,39(4):531-544. [22] 李蕾,邢昊,胡家,等.蛋白质磷酸化位点富集与鉴定方法的建立与应用[J].分析试验室,2015,34(11):1348-1352. [23] UBBIALI D, FRATINI M, PIERSIMONI L, et al. Direct observation of “elongated” conformational states in α-synuclein upon liquid-liquid phase separation. Angew Chem Int Ed Engl. 2022;61(46):e202205726. [24] KAWAHATA I, FINKELSTEIN DI, FUKUNAGA K. Pathogenic impact of α-synuclein phosphorylation and its kinases in α-synucleinopathies. Int J Mol Sci. 2022;23(11): 6216. [25] ANDERSON JP, WALKER DE, GODSTEIN JM, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739-29752. [26] VISANJI NP, WISLER-GENDEBIEN S, OSCHIPOK LW, et al. Withdrawal: effect of Ser-129 phosphorylation on interaction of α-synuclein with synaptic and cellular membranes [retraction of: J Biol Chem. 2011;286(41):35863-73]. J Biol Chem. 2020;295(39):13695. [27] GABRILIYAN L, LIANG H, MINALYAN A, et al. Behavioral deficits and brain α-synuclein and phosphorylated serine-129 α-synuclein in male and female mice overexpressing human α-synuclein. J Alzheimers Dis. 2021;79(2):875-893. [28] DING J, WANG Y, HUANG J, et al. Role of alpha-synuclein phosphorylation at Serine 129 in methamphetamine-induced neurotoxicity in vitro and in vivo. Neuroreport. 2020;31(11):787-797. [29] GHANEM SIMONA S, MAJBOUR NOUR K, VAIKATH NISHANT N, et al. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc Natl Acad Sci U S A. 2022;119: e2109617119. [30] VUIDEL A, COUSIN L, WEYKOPF B, et al. High-content phenotyping of Parkinson’s disease patient stem cell-derived midbrain dopaminergic neurons using machine learning classification. Stem Cell Reports. 2022;17(10):2349-2364. [31] CARIULO C, MARTUFI P, VERANI M, et al. Phospho-S129 alpha-synuclein is present in human plasma but not in cerebrospinal fluid as determined by an ultrasensitive immunoassay. Front Neurosci. 2019;13:889. [32] ARLINGHAUS R, IBA M, MASLIASH E, et al. Specific detection of physiological s129 phosphorylated α-synuclein in tissue using proximity ligation assay. J Parkinsons Dis. 2023;13(2):255-270. [33] WU W, SUNG CC, YU P, et al. Correction: S-nitrosylation of G protein-coupled receptor kinase 6 and Casein kinase 2 alpha modulates their kinase activity toward alpha-synuclein phosphorylation in an animal model of Parkinson’s disease. PLoS One. 2020;15(6):e0235296. [34] SANO K, IWASAKI Y, YAMASHITA Y, et al. Tyrosine 136 phosphorylation of α-synuclein aggregates in the Lewy body dementia brain: involvement of serine 129 phosphorylation by casein kinase 2. Acta Neuropathol Commun. 2021;9(1):182. [35] KEDARITI M, FRATTINI E, BADEN P, et al. LRRK2 kinase activity regulates GCase level and enzymatic activity differently depending on cell type in Parkinson’s disease. NP J Parkinsons Dis. 2022;8(1):92. [36] VERBINNEN I, VANEYNDE P, REYNHOUT S, et al. Protein Phosphatase 2A (PP2A) mutations in brain function, development, and neurologic disease. Biochem Soc Trans. 2021;49(4):1567-1588. [37] RUVOLO PP, DENG X, ITO T, et al. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274(29):20296-20300. [38] 安俊言,安思训,王鹏.帕金森病患者血清polo样激酶2活性升高促进α-突触核蛋白聚集[J].吉林医药学院学报,2019,40(3):171-174. [39] TOMA-FUKAI S, SHIMIZU T. Structural diversity of ubiquitin E3 ligase. Molecules. 2021;26(21):6682. [40] MOON SP, BALANA AT, GALESIC A, et al. Ubiquitination can change the structure of the α-synuclein amyloid fiber in a site selective fashion. J Org Chem. 2020;85(3): 1548-1555. [41] MEIER F, ABEYWARDANA T, DHALL A, et al. Semisynthetic,site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J Am Chem Soc. 2012;134(12):5468-5471. [42] WANG QJ, CHEN AD, ChEN HC, et al. Noncanonical roles of h α-syn (A53T) in the pathogenesis of parkinson’s disease: synaptic pathology and neuronal aging. Neural Plast. 2020(4):1-17. [43] 王洪财.P75介导的泛素化修饰在帕金森病α-突触核蛋白聚集中的调控机制[D].滨州:滨州医学院,2020. [44] BEHL T, KUMAR S, ALTHAFAR ZM, et al. Exploring the role of ubiquitin-proteasome system in Parkinson’s disease. Mol Neurobiol. 2022;59(7):4257-4273. [45] QUINN PMJ, MOREIRA PI, AMBROSIO AF, et al. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun. 2020; 8(1):189. [46] PARK SS, DO HA, PARK HB, et al. Deubiquitinating enzyme YOD1 deubiquitinates and destabilizes α-synuclein. Biochem Biophys Res Commun. 2023;645:124-131. [47] XU L, BHATTACHARYA S, THOMPSON D. On the ubiquity of helical α-synuclein tetramers. Phys Chem Chem Phys. 2019;21(22):12036-12043. [48] WATSON MD, LEE JC. N-terminal acetylation affects α-synuclein fibril polymorphism. Biochemistry. 2019;58(35):3630-3633. [49] BELL R, THRUSH RJ, CASTELLANA-CRUZ M, et al. N-terminal acetylation of α-synuclein slows down its aggregation process and alters the morphology of the resulting aggregates. Biochemistry. 2022;61(17):1743-1756. [50] 翟紫凝,吴琼,李从刚.乙酰化修饰抑制α-synuclein的纤维化聚集(英文)[J].波谱学杂志,2016,33(2):179-187. [51] YANG X, WANG B, HOOP CL, et al. NMR unveils an N-terminal interaction interface on acetylated-α-synuclein monomers for recruitment to fibrils. Proc Natl Acad Sci U S A. 2021;118(18):e2017452118. [52] KANG L, JANOWSKA MK, MORIARTY GM, et al. Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence. PLoS One. 2013;8(9):e75018. [53] TENG X, SHEVELEVA A, TUNA F, et al. Acetylation rather than H50Q mutation impacts the kinetics of Cu(II) binding to α-synuclein. Chemphyschem. 2021;22(23):2413-2419. [54] XIE Y, LUO X, LI Y. DeepNitro: prediction of protein nitration and nitrosylation sites by deep learning. Genomics, Proteomics Bioinformatics. 2018;16:294-306 [55] QIAO HH, ZHU LN, WANG Y, et al. Implications of alpha-synuclein nitration at tyrosine 39 in methamphetamine-induced neurotoxicity in vitro and in vivo. Neural Regen Res. 2019;14(2):319-327. [56] LIU Y, QIANG M, WEI Y, et al. A novel molecular mechanism for nitrated {alpha}-synuclein-induced cell death. J Mol Cell Biol. 2011;3(4):239-249. [57] MUSGROVE RE, HELWIG M, BAE EJ, et al. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J Clin Invest. 2019; 129(9):3738-3753. [58] STYKEL MG ,HUMPHRIES K, KIRBY MP, et al. Nitration of microtubules blocks axonal mitochondrial transport in a human pluripotent stem cell model of Parkinson’s disease. FASEB J. 2018;32:5350-5364. [59] GIASSON BI, DUDA JE, MURRAY IVJ, et al. Oxidative damage linked to neurodegeneration by selective a-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985-989. [60] SANTOS-LOBATO BL, BORTOLANZA M, PINHEIRO LC, et al. Levodopa-induced dyskinesias in Parkinson’s disease increase cerebrospinal fluid nitric oxide metabolites’ levels. J Neural Transm (Vienna). 2022;129:55-63. [61] MA LY, GAO LY, Li X, et al. Nitrated alpha-synuclein in minor salivary gland biopsies in Parkinson’s disease. Neurosci Lett. 2019;704:45-49. |
| [1] | Lou Guo, Zhang Yan, Fu Changxi. Role of endothelial nitric oxide synthase in exercise preconditioning-induced improvement of myocardial ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1283-1288. |
| [2] | Xie Peng, Zhang Jiang, Deng Xiaolei, Wei Bo, Hou Decai. A systematic review of mouse model construction for sarcopenia [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(2): 263-266. |
| [3] | Cheng Haotian, Zhao Xiaofeng, Lu Xiangdong, Zhao Yibo, Fan Zhifeng, Qi Detai, Wang Xiaonan, Zhou Runtian, Jin Xinjie, Zhao Bin. Single-cell RNA sequencing and the pathogenesis of intervertebral disc degeneration [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(1): 93-99. |
| [4] | Song Hehua, Wei Zairong. Diabetic peripheral neuropathy: research and therapy [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1278-1285. |
| [5] | Qi Xue, Li Jiahui, Zhu Yuanfeng, Yu Lu, Wang Peng. Phosphorylation modification of alpha synuclein in serum of patients with Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(35): 5577-5582. |
| [6] | Han Weiyu, Chen Yuanxing, Huang Youyang, Liu Weiwei, Zhao Yongchao, Zhao Ranzun. Regulation of cardiovascular diseases by histone deacetylation modification [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(35): 5707-5713. |
| [7] | Zheng Xiaohan, Feng Xiaoli, Hu Lan, Gao Shijun, Wei Yanzhao, Huang Ting, Sun Shengtong, Wei Xufang, Wang Tan, Zhao Zhenqiang. Macrophage migration inhibitory factor promotes the differentiation of human embryonic stem cells into ventral midbrain dopaminergic neural progenitor cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(33): 5348-5356. |
| [8] | Wang Ziyang, Zuo Enjun. Application of biological materials in vital pulp therapy [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(3): 427-433. |
| [9] | Wang Yiying, Li Ruiqing, Li Jingwen, Mei Jinjin, Zhang Jianyun, Zhang Lihong, Fan Yongfu, Guo Jian. Exosome-mediated cellular communication: a potential biomarker for Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(24): 3883-3891. |
| [10] | Li Yangjie, Qi Rong, Zhang Xinyu, Cheng Jiaji, Zhou Ning, Cui Xue, Cheng Shuang, Wang Zhengdong, Yan Nan. Neuroprotective effects of sodium butyrate and acetylation proteomics analysis in fluorosis rats [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(20): 3151-3157. |
| [11] | Zhang Jian, Lin Jianping, Zhou Gang, Wang Benchao, Wu Yongchang. Etiological analysis of discoid meniscus based on whole exome sequencing [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(2): 192-199. |
| [12] | Tang Yujing, Lan Fengjun, Li Guangdi, Wang Jian, Liu Riguang. Role of calcium ions in the pathogenesis of chronic fluorosis [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(17): 2745-2753. |
| [13] | He Zike, Wang Shangzeng. Eucommia ulmoides Oliver aqueous extract promotes bone marrow mesenchymal stem cell proliferation and osteoblastic differentiation through upregulating Nur77 protein expression [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(15): 2371-2378. |
| [14] | Chen Shijian, Li Ge, Zhang Yu, Guan Yalun, Li Xuejiao, Liu Shuhua, Li Yongchao, Li Yunfeng, Gao Jinfeng, Wei Xiaoyue, Zhao Yuhong. Comparison and evaluation of MPTP-induced subacute and chronic models of Parkinson’s disease in mice [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1247-1252. |
| [15] | Xiao Yang, Gong Liqiong, Fei Jing, Li Leiji. Effect of electroacupuncture on nerve growth factor and its receptor expression in facial nerve nucleus after facial nerve injury in rabbits [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1253-1259. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||