Chinese Journal of Tissue Engineering Research ›› 2023, Vol. 27 ›› Issue (34): 5561-5569.doi: 10.12307/2023.717
Previous Articles Next Articles
Signaling pathway of osteoblast autophagy in periprosthetic osteolysis
Gu Yingchu1, Gu Ye1, Wu Zerui1, Fang Tao1, Wang Qiufei1, Chen Bingqian1, Peng Yuqin1, Geng Dechun2, Xu Yaozeng2
- 1Department of Orthopedics, Changshu First People’s Hospital, Affiliated Changshu Hospital of Soochow University, Changshu 215500, Jiangsu Province, China; 2Department of Orthopedics, First Affiliated Hospital of Soochow University, Suzhou 215006, Jiangsu Province, China
-
Received:2022-10-12Accepted:2022-11-25Online:2023-12-08Published:2023-04-23 -
Contact:Gu Ye, MD, Associate chief physician, Department of Orthopedics, Changshu First People’s Hospital, Affiliated Changshu Hospital of Soochow University, Changshu 215500, Jiangsu Province, China -
About author:Gu Yingchu, Master, Physician, Department of Orthopedics, Changshu First People’s Hospital, Affiliated Changshu Hospital of Soochow University, Changshu 215500, Jiangsu Province, China -
Supported by:Youth Medical Key Talent Funding Project of Jiangsu Province, No. QNRC2016751 (to GDC); Six-One Engineering Funded Project of Jiangsu Province, No. LQY2016033 (to GDC); Key Research and Development Program of Jiangsu Provincial Science and Technology Department (Social Development) Project, No. BE2021673, BE2020666 (to GY); Science and Technology Development Program of Suzhou Science and Technology Bureau, No. SYSD2022023, SYSD2020013 (to GY); Clinical Diagnosis and Treatment of Key Diseases of Suzhou Municipal Health Commission, No. LCZX201824 (to GY); Science and Technology Development Project of Science and Technology Bureau of Changshu City of Jiangsu Province, No. CS202119 (to WQF), No. CS201817 (to GY); Horizontal Project of Soochow University, No. H200833 (to GY)
CLC Number:
Cite this article
Gu Yingchu, Gu Ye, Wu Zerui, Fang Tao, Wang Qiufei, Chen Bingqian, Peng Yuqin, Geng Dechun, Xu Yaozeng. Signaling pathway of osteoblast autophagy in periprosthetic osteolysis[J]. Chinese Journal of Tissue Engineering Research, 2023, 27(34): 5561-5569.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks

为了进一步了解假体周围骨溶解中成骨细胞自噬的机制,文章总结了调控自噬的信号通路或关键基因,并对它们进行了分类并简述了其主要作用机制,后续将以此展开讨论并论述其在假体周围骨溶解中的作用,见表3。"

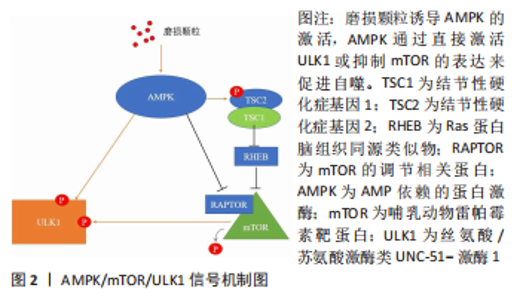
2.1 AMPK/mTOR/ULK1信号通路动态调控成骨细胞自噬水平 AMPK即 AMP依赖的蛋白激酶[Adenosine 5’-monophosphate (AMP)-activated protein kinase,AMPK],它是由α、β和γ亚基组成异源三聚体复合物。AMPK 活化可提高分解代谢(ATP 产生)途径的速率,并降低合成代谢(ATP 利用)途径的速率。通过这种方式,它在全身能量代谢中起着重要作用[25]。在过去几年中,AMPK 已被确定为线粒体稳态的守护者[26]。AMPK激活可通过两种不同机制诱导自噬过程:抑制哺乳动物雷帕霉素靶蛋白(mechanistic target of rapamycin kinase,mTOR)和哺乳动物自噬起始激酶(unc-51 like autophagy activating kinase 1,ULK1)的直接磷酸化,并且mTOR与 ULK1之间也存在着相互作用关系,因此AMPK,mTOR及ULK1可以如三角形般互相激活或抑制[27]。当能量水平较低或存在饥饿情况时,AMPK被磷酸化,激活的AMPK通过两种方式抑制mTOR的活性:①AMPK在其T1227和S1345位点直接磷酸化TSC2,这促进了TSC1/TSC2复合物的GTP酶活性。TSC1/TSC2复合物能水解GTP并使RHEB失活,导致RHEB失去介导mTORC1激活的能力。②AMPK直接磷酸化RAPTOR的Ser772和Ser792位点,阻碍RAPTOR与mTOR或mTOR底物的结合,进而抑制mTOR信号通路。mTOR的失活消除了其对ULK1在Ser757位点的磷酸化抑制,并诱导了ULK1与AMPK的结合,这是AMPK增加自噬的主要机制之一[28]。除此以外,AMPK可以直接磷酸化ULK1多个位点,以此增加自噬活性[29]。总之,AMPK与mTOR均可以调控ULK1,累积的报告表明,AMPK,mTOR和ULK1之间存在双向调节,这意味着AMPK,ULK1和mTOR形成了一个信号三元组,它们通过互相调节以维持自噬的动态平衡[30]。AMPK/mTOR/ULK1信号通路的机制,见图2。"
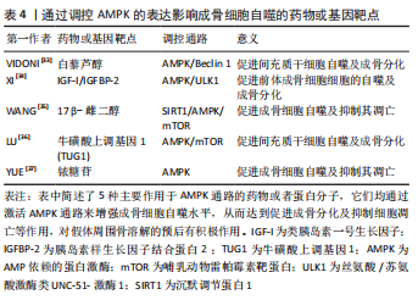
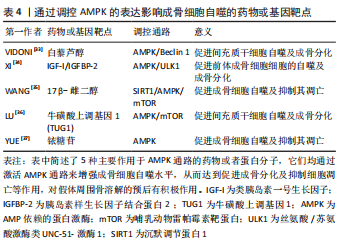
AMPK 调控自噬的能力在骨溶解疾病中也能对成骨细胞的功能与活性带来影响。伤口部位的负压引流可以通过激活AMPK和调节ULK1来增强成骨细胞自噬,从而促进体外和体内的骨再生[31]。在人牙髓间充质干细胞的成骨分化中,AMPK的敲低阻止了早期mTOR抑制并减弱了自噬水平,阻碍了成骨细胞的分化。而Akt的抑制可以减少mTOR活化并增强自噬水平,促进成骨细胞分化[32]。除此以外,还有许多通过调控AMPK表达来影响成骨细胞自噬的药物或基因靶点已被发现[33-37],它们在下方表格中被列出,见表4。"

生理条件下,成骨细胞的自噬维持在基础水平,当它面临过量的有害刺激如磨损颗粒、氧化应激、炎症等时会提高自身自噬水平以实现自我保护。然而,大部分情况下成骨细胞的自噬水平不足以对抗外来刺激,最终导致骨溶解的发生。AMPK信号通路是磨损颗粒诱导的成骨细胞自噬水平提高的主要途径之一,AMPK的上调会导致mTOR的抑制和ULK1的活化,这两者都会提高成骨细胞的自噬能力,从而提高成骨细胞面对有害刺激时的自我保护能力。因此,靶向激活AMPK促进自噬已成为治疗假体周围骨质溶解的潜在措施之一。"

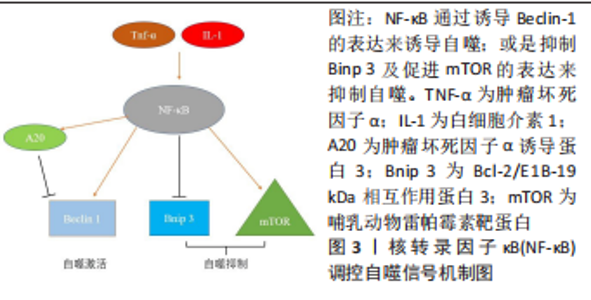
2.2 核转录因子κB信号通路与成骨细胞自噬之间存在串扰 核转录因子κB家族成员与反转录病毒癌蛋白v-Rel统归为核转录因子κB/Rel蛋白家族,它们在炎症和免疫反应中起重要作用[38]。核转录因子κB通过5个亚单位的二聚化形成,包括核转录因子κB1 (p50),核转录因子κB2(p52),RELA(也称为p65),RELB和c-REL[39]。核转录因子κB激活通过两种主要信号通路发生:经典和非经典核转录因子κB信号传导。经典途径介导核转录因子κB1 p50,RELA和c-Rel的激活,而非经典核转录因子κB途径选择性激活核转录因子κB2 p52和RELB[40]。基于白细胞介素1和肿瘤坏死因子α等促炎因子激活核转录因子κB以及核转录因子κB对其他促炎基因的影响,核转录因子κB被认为是典型的促炎信号通路[41]。同时,核转录因子κB的抗凋亡作用使其在肿瘤的发生和发展中起着至关重要的作用[42]。最近的研究表明,核转录因子κB信号通路和自噬之间存在复杂的串扰[43]。自噬由核转录因子κB通过其诱导物产生正向或负向控制,在尤因肉瘤、乳腺癌和白血病细胞中,肿瘤坏死因子介导的核转录因子κB激活通过活性氧抑制和mTOR激活抑制自噬[44]。 在缺氧条件下,缺氧诱导因子1α通过核转录因子κB诱导自噬[45]。在热休克应激下,IκB/核转录因子κB复合物失去稳定性并激活核转录因子κB[46]。核转录因子κB还可以通过调节自噬相关基因的表达和活性来影响自噬。核转录因子κB及其家族成员p65/RelA识别并结合Beclin-1的启动子,促进Beclin-1基因表达并增强自噬。有趣的是,核转录因子κB对Beclin-1的调节也具有相反的效果。核转录因子κB 激活上调 A20 的表达,A20 是一种抑制 Beclin-1 泛素化从而限制自噬的因子[47]。此外,核转录因子κB通过调节Bnip3来调节自噬。正常情况下,p65阻止E2F1与Bnip3启动子结合,从而抑制Bnip3的转录并降低自噬水平。缺氧时核转录因子κB活性降低,导致Bnip3表达水平增加,并诱导自噬[48]。图3给出了核转录因子κB调控自噬的部分信号传导通路。"
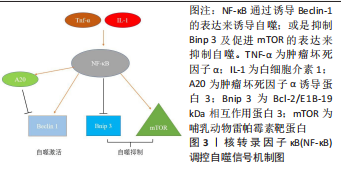

一些研究指出,核转录因子κB信号通路可促进成骨细胞自噬,从而在调节成骨细胞分化中起重要作用[49]。无需药物刺激或其他干预,成骨细胞自身具一定的基础自噬水平,SN50作为核转录因子κB通路的阻断剂,只有在一定的质量浓度(25 mg/mL)对自噬起到显著作用,通过阻断核转录因子κB通路可以显著抑制成骨细胞的自噬水平[50]。肿瘤坏死因子α通过激活P38 MAPK/核转录因子κB通路,可促进成骨细胞自噬,减少股骨头缺血性坏死的成骨细胞凋亡[51]。低浓度的脂多糖通过成骨细胞中的核转录因子κB信号通路激活自噬并促进细胞增殖,从而抑制细胞凋亡,加速骨折愈合[52]。然而,相反的研究表明薯蓣皂苷通过ASPP2/核转录因子κB 通路抑制成骨细胞自噬来增强成骨细胞的分化和增殖能力[53]。 核转录因子κB作为经典的炎症通路与自噬之间的存在复杂的串扰,不同条件下对于核转录因子κB的干预可能会为成骨细胞的活性及功能带来不同的影响,并且核转录因子κB可以调控多种自噬相关基因的表达,核转录因子κB通路与细胞自噬关系密切。核转录因子κB 对自噬的调控因此提供了将炎症和免疫信号传导与细胞死亡和自我吞噬联系起来的机制,但目前关于此方面的研究还不充分,将核转录因子κB作为骨溶解疾病的治疗靶点还需要更多的研究证据来阐明其中的潜在机制。"


2.3 FoxO3 通过调控自噬水平来应对成骨细胞氧化应激损伤 在哺乳动物中,FoxO家族共有4位成员,FoxO1,FoxO3,FoxO4和FoxO6。FoxOs最初被确定为胰岛素通路的下游调节因子,已知可与多种靶基因的启动子结合,并控制对细胞稳态至关重要的几个过程,包括细胞能量产生、抗氧化应激和细胞活力和增殖[54]。FoxO转录因子的活性可被胰岛素和生长因子信号抑制。在存在胰岛素和胰岛素样生长因子的情况下,PI3K-Akt信号通路被激活,激酶如Akt和SGK在3个保守残基处直接磷酸化FoxO因子,导致FoxO结合细胞质蛋白14-3-3并抑制其转录活性。而在缺乏胰岛素或生长因子信号的情况下,或在饥饿期间,FoxO因子转移到细胞核,在那里它们激活一些应激反应或代谢基因的表达[55]。虽然FoxO家族均有诱导自噬的能力,但其中FoxO3的作用最为重要。FoxO3长期以来一直被认为在长寿的分子基础中起着关键作用[56]。FoxO3在饥饿时易位到细胞核中,然后诱导参与该过程各个阶段的许多自噬基因的表达,包括LC3b,Gabarapl1,PI3KII,Ulk1,Atg12,Beclin-1,Atg4b和Bnip3,以促进自噬。Toll样受体4 (TLR4)通过PI3K-FoxO3途径抑制小胶质细胞的自噬,从而降低其吞噬能力[57]。 最近,有报道称在成骨细胞中FoxO3可以通过调节自噬来影响细胞凋亡[58]。在间充质干细胞中,需要激活FoxO3以诱导自噬,从而减少成骨细胞分化过程中线粒体呼吸增加引起的活性氧水平升高并减少成骨细胞凋亡。有学者研究了人骨髓间充质干细胞的成骨分化,发现FoxO3被活性氧激活,这是由线粒体代谢增加引起的,以获得足够的分化能量。该过程主要依赖于丝裂原活化蛋白激酶8(MAPK8)诱导的FoxO3的ser294磷酸化,因此,FoxO3活化对于通过激活自噬来调控活性氧水平很重要[59]。 氧化应激损伤导致的成骨细胞内活性氧增加会诱导FoxO3的激活,而激活的FoxO3可以诱导多种自噬基因的表达,成骨细胞自噬会清除损伤的细胞器并回收能量以减少成骨细胞凋亡,而FoxO3的敲除会抑制细胞的自我净化能力。但是,当氧化应激损伤诱导的活性氧超过了FoxO3激活所带来的自我净化效果,则无法阻止细胞凋亡。在假体周围骨溶解中,磨损粒子诱导的氧化应激损伤在发病机制中占有重要地位,因此,靶向FoxO3从而激活自噬的治疗方法可能对假体周围骨溶解的治疗与预后带来一定的改善。"


2.4 泛素化货物的运输者——p62 p62(sqstm1)是一种泛素结合蛋白,参与细胞信号转导、氧化应激和自噬。Komatsu等[60]在2007 年报告了p62和LC3之间的关联,表明p62在自噬中的作用。p62是一种多功能蛋白,它由N端Phox-BEM1结构域(PB1),锌型锌指结构域,核定位信号(NLS),输出基序(NES),LC3相互作用区(LIR),Keap1相互作用区(KIR)和C端泛素相关结构域(UBA)组成[61]。p62通过C端UBA结构域或LIR结构域输送泛素货物至溶酶体以进行降解[62]。在自噬过程中,溶酶体自噬的降解导致sqstm1水平的降低。相反,自噬抑制蛋白可以稳定sqstm1水平[63]。现在可以理解的是,p62 作为一个适配器发挥作用,结合泛素化的蛋白质聚集体并将它们传递给溶酶体。P62的转录由各种信号通路介导,如氧化应激(Nrf2),Ras/MAPK及JNK/c-Jun等[64]。最近的研究表明,成骨细胞自噬可以通过调节p62来介导。大麻素受体2型(CNR2)可以通过激活p62介导的自噬来诱导成骨分化[65]。在2型糖尿病大鼠中,运动可以下调长链非编码RNA meg3(meg3)并激活p62介导的自噬以促进骨形成[66]。p62可以通过KEAP1-NrF2途径参与泛素-蛋白酶体系统(UPS)的调节[67]。p62是各种细胞功能的整合中心,包括自噬体的形成和泛素化蛋白质向溶酶体的传递。p62作为自噬过程中的关键基因之一值得进一步研究。"

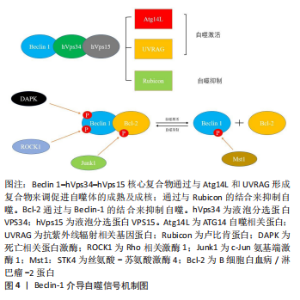
2.5 Beclin-1——自噬体形成与成熟的关键参与者 Beclin-1通过与不同蛋白质形成复合物来调节自噬,这些研究表明,Beclin-1和Ⅲ类PI3K(hVps34,hVps15)形成复合物[68]。Beclin 1–hVps34–hVps15核心复合物通过与Atg14L,UVRAG和Rubicon形成不同复合物来调节自噬的启动和内吞途径。Beclin 1–hVps34–hVps15-Atg14L有助于自噬体成核,Beclin 1–hVps34–hVps15-UVRAG有助于自噬体成熟,Beclin 1–hVps34–hVps15-Rubicon则会抑制自噬[69]。除此以外,Beclin 1与Bcl-2的结合也对自噬起着调控作用。Bcl-2/Bcl-xl与Beclin 1的结合会抑制细胞自噬,生理状态下,Bcl-2/Bcl-xl可以通过其BH3受体结构域与Beclin-1的BH3结构域结合,Bcl-2–Beclin 1复合物的动态相互作用在正常范围内调节自噬,以维持体内平衡[70]。自噬基因Beclin-1的缺失将自噬活性降低到异常水平,导致营养缺乏和其他应激条件下的细胞死亡;相反,Bcl-2的缺失诱导自噬过度活性,这也可能导致细胞死亡。 c-Jun N端蛋白激酶1(JNK1)可以磷酸化Bcl-2,解离Bcl 2–Beclin 1复合物并诱导自噬[71]。死亡诱导激酶(DAPK)的过度表达会使Beclin-1的BH3结构域的苏氨酸119位点磷酸化,并促进Becli-1和Bcl-xl的解离,从而诱导自噬[72]。ROCK1磷酸化Beclin-1的T119位点,并导致其与Bcl-2分离。ROCK1基因敲除小鼠的自噬诱导受损。这些结果表明,ROCK1是Beclin-1上游的关键调节器[73]。Mst1在应激下诱导并磷酸化Beclin-1的BH3结构域的T108位点。与T119的磷酸化相反,T108的磷酸化增强了Beclin-1和Bcl-2/Bcl-xl之间的相互作用,并稳定了Beclin-1同二聚体,这抑制了Atg14L–Beclin-1–Vps34复合物的PI3K激酶活性,并最终抑制自噬[74]。Beclin-1复合物的组成及其与Bcl-2之间的调控机制见图4。"
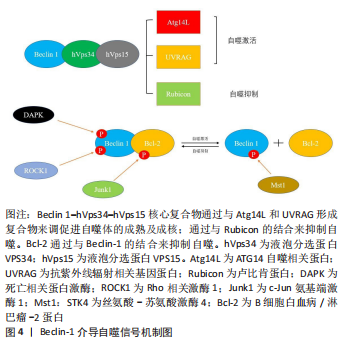

Beclin-1因其在自噬过程中的关键作用被鉴定为自噬标志性基因,在成骨细胞的自噬中也是如此。PTH1-34可通过调控Beclin-1的表达来增强成骨细胞的自噬活性,从而改善成骨细胞的增殖和矿化能力,减少骨丢失[75]。目前许多调控成骨细胞自噬的药物在使用后均观察到了Beclin-1的表达变化。白藜芦醇治疗可显著改善骨质疏松大鼠的骨质量并降低血清碱性磷酸酶和骨钙素水平。有研究显示,高剂量白藜芦醇治疗的骨质疏松大鼠成骨细胞中 Beclin-1 的表达增加,并且白藜芦醇处理增加了地塞米松处理的成骨细胞中 Beclin-1 mRNA 表达,细胞自噬增加且凋亡受到抑制[76]。 综上所述,生理情况下,Beclin-1与Bcl-2动态结合的平衡是维系自噬稳态的重要因素,磨损颗粒诱导的过度自噬可能通过调控Beclin-1与Bcl-2的磷酸化水平得以改善,并且Beclin-1是自噬体成熟及成核的关键组成部分,因此通过干预Beclin-1的表达对成骨细胞自噬能力进行调控,可能会对假体周围骨溶解的治疗有重要意义。"


2.7 PP2AC——成骨细胞自噬的潜在调控基因 蛋白质磷酸酶2A(protein phosphatase 2A,PP2A)是一种丝氨酸-苏氨酸磷酸酶,可调节体内磷酸化[83]。同时,PP2A是一种三聚体,由催化亚基C、结构亚基A和调控亚基B组成,其功能主要取决于催化亚基C的调控[84]。在此基础上,PP2A全酶组合物的多样性为其在细胞内的调节作用提供了多种可能性。以前的文献已经证实它对肿瘤、呼吸系统疾病和神经系统疾病有重要作用。PP2Ac甲基化水平的调节可以通过mTORC1/ULK1信号通路改善MN2a细胞中MN诱导的异常自噬[85]。PP2Ac在牛分枝杆菌感染的巨噬细胞中表达显著增加[86]。尼罗替尼抑制PP2Ac表达后,可通过AMPK信号通路在巨噬细胞中诱导保护性自噬,从而提高细胞的抗菌能力和存活能力[87]。牛磺酸通过抑制PP2Ac甲基化来阻断线粒体自旋,从而减少巨噬细胞过度的促炎极化。VDAC1和PINK1是参与PP2Ac甲基化介导的线粒体自大的介质,是维持PP2Ac高甲基化所必需的[88]。近年来,许多学者发现PP2A也与骨科疾病有关。OKAMURA等[88]发现,PP2A的减少通过调节Osterix和其他与骨相关的转录因子来促进成骨细胞的分化。PP2Ac与多种自噬信号通路密切相关,如mTOR/ULK1,PINK1/Parkin和AMPK等,并且能调节成骨细胞的分化和功能。虽然目前还缺乏PP2Ac通过调控成骨细胞自噬干预其生理活性及功能的直接证据,但PP2Ac作为假体周围骨溶解的潜在作用靶点值得进一步深入研究。"


2.8 PINK1/Parkin——线粒体自噬的经典途径 除了非选择性自噬,还有选择性自噬,如线粒体自噬,其是通过以选择性自噬形式降解线粒体,调节线粒体的质量和数量控制。通过特定的线粒体外膜受体或与线粒体表面蛋白质结合的泛素分子促进线粒体自噬,从而在线粒体周围形成自噬体[89]。线粒体自噬介导的线粒体消除在包括早期胚胎发育、细胞分化、炎症和细胞凋亡在内的许多过程中发挥着重要作用。线粒体自噬以“细胞内管家”的身份对于细胞的增殖分化起到了保护作用[90]。 Parkin是一种由465个氨基酸组成的蛋白质,基因长度为1.4 MB,是仅次于抗营养蛋白的第二大人类基因,它具有氨基末端泛素结构域和羧基末端环盒。1998年,Parkin被确定为常染色体隐性青少年帕金森病(AR-JP)的致病基因[91]。PINK1是早发性帕金森病的另一个致病基因,与AR-JP一样,以常染色体隐性遗传的方式遗传[92],虽然它们具有相同的致病作用,但它们编码的蛋白质具有完全不同的分子功能:一个是蛋白激酶,另一个是泛素连接酶。PINK1/Parkin作为一种经典的线粒体自律途径已被学者广泛研究[93]。PINK1作为一名检查员平时持续监测线粒体健康,并在线粒体功能受损时迅速做出反应。在健康线粒体中,PINK1在穿过线粒体膜的过程中会被泛素-蛋白酶体系统快速降解。相反,受损线粒体由于失去维持跨膜电位的能力而导致PINK1的积累,导致快速的Parkin募集和激活,随后启动线粒体自噬[94]。在生理状态下,Parkin通过多个分子内相互作用保持在“封闭”状态,使酶处于自动抑制状态,PINK1通过直接磷酸化Parkin的Ubl结构域中的S65位点来激活Parkin[95]。PINK1/Parkin的缺失在帕金森病的发生和发展中起着重要作用。Parkin和PINK突变都显示出线粒体蛋白更新和线粒体自噬的显著抑制。未能去除受损的线粒体蛋白在帕金森病的发病机制中起重要作用[96]。 近年来研究称,PINK1/Parkin通路对于成骨细胞的功能也有很重要的影响,Parkin通过激活β-连环蛋白信号通路来增强自噬并促进骨髓间充质干细胞的成骨细胞分化;相反,Parkin表达的自身调节导致更严重的成骨分化,如间充质干细胞脂肪变性和Ⅰ型胶原表达减少[97]。VK2可以通过上调Parkin和其他自噬和线粒体标志物的表达来逆转地塞米松诱导的自噬抑制,从而改善成骨细胞功能[98]。 线粒体作为细胞的“动力源”主要负责合成能量及介导一系列细胞内过程。线粒体自噬是细胞维护自身正常功能、维系细胞稳态的重要生理过程,PINK1/Parkin通路在其中扮演着重要角色。假体周围骨溶解中成骨细胞的凋亡原因之一即线粒体功能失调导致的线粒体膜去极化和细胞整体氧化应激增加,因此通过调控PINK1/Parkin通路发挥成骨细胞的线粒体自噬功能是其应对磨损颗粒损害的有效措施之一。PINK1/Parkin作为线粒体自噬的主要通路之一,其与假体周围骨溶解的关系值得进一步研究,并且应当更多地关注PINK1/Parkin通路与其他自噬通路之间的串扰。"

| [1] TALMO CT, ROBBINS CE, BONO JV. Total joint replacement in the elderly patient. Clin Geriatr Med. 2010;26(3):517-529. [2] SLOAN M, PREMKUMAR A, SHETH NP. Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. J Bone Joint Surg Am. 2018;100(17):1455-1460. [3] TALMO CT, AGHAZADEH M, BONO JV. Perioperative complications following total joint replacement. Clin Geriatr Med. 2012;28(3):471-487. [4] PURDUE PE, KOULOUVARIS P, POTTER HG, et al. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res. 2007;454:251-261. [5] BLOM AW, HUNT LP, MATHARU GS, et al. The effect of surgical approach in total knee replacement on outcomes. An analysis of 875, 166 elective operations from the National Joint Registry for England, Wales, Northern Ireland and the Isle of Man. Knee. 2021;31:144-157. [6] OLLIVERE B, WIMHURST JA, CLARK IM, et al. Current concepts in osteolysis. J Bone Joint Surg Br. 2012;94(1):10-15. [7] NOORDIN S, MASRI B. Periprosthetic osteolysis: genetics, mechanisms and potential therapeutic interventions. Can J Surg. 2012;55(6):408-417. [8] KIM JM, LIN C, STAVRE Z, et al. Osteoblast-osteoclast communication and bone homeostasis. Cells. 2020;9(9):2073. [9] VERMES C, GLANT TT, HALLAB NJ, et al. The potential role of the osteoblast in the development of periprosthetic osteolysis: review of in vitro osteoblast responses to wear debris, corrosion products, and cytokines and growth factors. J Arthroplasty. 2001;16(8 Suppl 1):95-100. [10] O’NEILL SC, QUEALLY JM, DEVITT BM, et al. The role of osteoblasts in peri-prosthetic osteolysis. Bone Joint J. 2013;95-B(8):1022-1026. [11] DIKIC I, ELAZAR Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349-364. [12] KIM KH, LEE MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322-337. [13] MAIURI MC, ZALCKVAR E, KIMCHI A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007; 8(9):741-752. [14] MIZUSHIMA N, LEVINE B, CUERVO AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069-1075. [15] DI GIACOMO V, CATALDI A, SANCILIO S. Biological factors, metals, and biomaterials regulating osteogenesis through autophagy. Int J Mol Sci. 2020; 21(8):2789. [16] MA B, GUAN G, LV Q, et al. Curcumin ameliorates palmitic acid-induced saos-2 cell apoptosis via inhibiting oxidative stress and autophagy. Evid Based Complement Alternat Med. 2021;2021:5563660. [17] 蔡燕,施勤,赵环,等.聚甲基丙烯酸甲酯颗粒诱导骨溶解实验研究[J].重庆医学,2013,42(34):4160-4161,4165. [18] WANG Z, LIU N, LIU K, et al. Autophagy mediated CoCrMo particle-induced peri-implant osteolysis by promoting osteoblast apoptosis. Autophagy. 2015;11(12):2358-2369. [19] LIU N, MENG J, WANG Z, et al. Autophagy mediated TiAl₆V₄ particle-induced peri-implant osteolysis by promoting expression of TNF-α. Biochem Biophys Res Commun. 2016;473(1):133-139. [20] WANG Z, DENG Z, GAN J, et al. TiAl6V4 particles promote osteoclast formation via autophagy-mediated downregulation of interferon-beta in osteocytes. Acta Biomater. 2017;48:489-498. [21] YAN JQ, ZHANG Y, LIU FS, et al. TCP wear particles causes injury of periprosthetic osteocytes in the mouse calvaria. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2018;34(1):83-87. [22] LI D, WANG C, LI Z, et al. Nano-sized AlO particle-induced autophagy reduces osteolysis in aseptic loosening of total hip arthroplasty by negative feedback regulation of RANKL expression in fibroblasts. Cell Death Dis. 2018;9(8):840. [23] 陈洋,宁乐,杜嘉莉,等.c-Jun氨基末端激酶介导的细胞凋亡和自噬参与调控磷酸三钙磨损颗粒诱导的小鼠颅骨假体周围骨细胞死亡[J].解剖学报,2018,49(5):605-610. [24] 林琨,陈佳濠,方泽浩,等.原花青素对磷酸三钙磨损颗粒所致小鼠颅骨溶解的干预作用及其机制[J].中国应用生理学杂志,2019,35(3):250-255. [25] CARLING D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017; 45:31-37. [26] FRIIS RMN, GLAVES JP, HUAN T, et al. Rewiring AMPK and mitochondrial retrograde signaling for metabolic control of aging and histone acetylation in respiratory-defective cells. Cell Rep. 2014;7(2):565-574. [27] ALERS S, LÖFFLER AS, WESSELBORG S, et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2-11. [28] LI Y, CHEN Y. AMPK and Autophagy. Adv Exp Med Biol. 2019;1206:85-108. [29] KIM J, KUNDU M, VIOLLET B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-141. [30] WONG PM, PUENTE C, GANLEY IG, et al. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124-137. [31] ZHANG S, XIE Y, YAN F, et al. Negative pressure wound therapy improves bone regeneration by promoting osteogenic differentiation via the AMPK-ULK1-autophagy axis. Autophagy. 2022;18(9):2229-2245. [32] PANTOVIC A, KRSTIC A, JANJETOVIC K, et al. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone. 2013; 52(1):524-531. [33] VIDONI C, FERRARESI A, SECOMANDI E, et al. Autophagy drives osteogenic differentiation of human gingival mesenchymal stem cells. Cell Commun Signal. 2019;17(1):98. [34] XI G, ROSEN CJ, CLEMMONS DR. IGF-I and IGFBP-2 stimulate AMPK activation and autophagy, which are required for osteoblast differentiation. Endocrinology. 2016;157(1):268-281. [35] WANG Y, MEI R, HAO S, et al. Up-regulation of SIRT1 induced by 17beta-estradiol promotes autophagy and inhibits apoptosis in osteoblasts. Aging (Albany NY). 2021;13(20):23652-23671. [36] LU DG, LU MJ, YAO SH, et al. Long non-coding RNA TUG1 promotes the osteogenic differentiation of bone marrow mesenchymal stem cells by regulating the AMPK/mTOR/autophagy pathway. Biomed Res. 2021;42(6): 239-246. [37] YUE C, JIN H, ZHANG X, et al. Aucubin prevents steroid-induced osteoblast apoptosis by enhancing autophagy via AMPK activation. J Cell Mol Med. 2021;25(21):10175-10184. [38] SUN SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545-558. [39] HAYDEN MS, GHOSH S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344-362. [40] SUN SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011;21(1):71-85. [41] LAWRENCE T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. [42] HOESEL B, SCHMID JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. [43] VERZELLA D, PESCATORE A, CAPECE D, et al. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020;11(3):210. [44] DJAVAHERI-MERGNY M, AMELOTTI M, MATHIEU J, et al. Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy. 2007;3(4):390-392. [45] MARTINEZ-OUTSCHOORN UE, WHITAKER-MENEZES D, PAVLIDES S, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: a simple solution to the autophagy paradox. Cell Cycle. 2010;9(21):4297-4306. [46] NIVON M, RICHET E, CODOGNO P, et al. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy. 2009;5(6):766-783. [47] TROCOLI A, DJAVAHERI-MERGNY M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am J Cancer Res. 2011;1(5):629-649. [48] SHAW J, YURKOVA N, ZHANG T, et al. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc Natl Acad Sci U S A. 2008;105(52):20734-20739. [49] ZHANG L, SUN Y, XU W, et al. Baicalin inhibits Salmonella typhimurium-induced inflammation and mediates autophagy through TLR4/MAPK/NF-κB signalling pathway. Basic Clin Pharmacol Toxicol. 2021;128(2):241-255. [50] QIN H, XU HZ, GONG YQ. Mechanism of NF-κB signaling pathway and autophagy in the regulation of osteoblast differentiation. Mol Membr Biol. 2016;33(6-8):138-144. [51] ZHENG LW, WANG WC, MAO XZ, et al. TNF-α regulates the early development of avascular necrosis of the femoral head by mediating osteoblast autophagy and apoptosis via the p38 MAPK/NF-κB signaling pathway. Cell Biol Int. 2020;44(9):1881-1889. [52] XU MX, SUN XX, LI W, et al. LPS at low concentration promotes the fracture healing through regulating the autophagy of osteoblasts via NF-κB signal pathway. Eur Rev Med Pharmacol Sci. 2018;22(6):1569-1579. [53] ZHU C, BAO N, CHEN S, et al. Dioscin enhances osteoblastic cell differentiation and proliferation by inhibiting cell autophagy via the ASPP2/NF-κβ pathway. Mol Med Rep. 2017;16(4):4922-4926. [54] LINK W. Introduction to FOXO Biology. Methods Mol Biol. 2019;1890:1-9. [55] BHARDWAJ G, PENNIMAN CM, JENA J, et al. Insulin and IGF-1 receptors regulate complex I-dependent mitochondrial bioenergetics and supercomplexes via FoxOs in muscle. J Clin Invest. 2021;131(18):e146415. [56] MORRIS BJ, WILLCOX DC, DONLON TA, et al. FOXO3: a major gene for human longevity--a mini-review. Gerontology. 2015;61(6):515-525. [57] LEE JW, NAM H, KIM LE, et al. TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy. 2019;15(5):753-770. [58] FITZWALTER BE, THORBURN A. FOXO3 links autophagy to apoptosis. Autophagy. 2018;14(8):1467-1468. [59] GÓMEZ-PUERTO MC, VERHAGEN LP, BRAAT AK, et al. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy. 2016;12(10):1804-1816. [60] KOMATSU M, WAGURI S, KOIKE M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149-1163. [61] LIN X, LI S, ZHAO Y, et al. Interaction domains of p62: a bridge between p62 and selective autophagy. DNA Cell Biol. 2013;32(5):220-227. [62] MYEKU N, FIGUEIREDO-PEREIRA ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286(25):22426-22440. [63] KATSURAGI Y, ICHIMURA Y, KOMATSU M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282(24):4672-4678. [64] PUISSANT A, FENOUILLE N, AUBERGER P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. 2012;2(4):397-413. [65] XU A, YANG Y, SHAO Y, et al. Activation of cannabinoid receptor type 2-induced osteogenic differentiation involves autophagy induction and p62-mediated Nrf2 deactivation. Cell Commun Signal. 2020;18(1):9. [66] CHEN X, YANG K, JIN X, et al. Bone autophagy: a potential way of exercise-mediated Meg3/P62/Runx2 pathway to regulate bone formation in T2DM mice. Diabetes Metab Syndr Obes. 2021;14:2753-2764. [67] LIU WJ, YE L, HUANG WF, et al. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. [68] HURLEY JH, YOUNG LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225-244. [69] XU HD, QIN ZH. Beclin 1, Bcl-2 and Autophagy. Adv Exp Med Biol. 2019; 1206:109-126. [70] KANG R, ZEH HJ, LOTZE MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571-580. [71] WEI Y, PATTINGRE S, SINHA S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678-688. [72] LEVIN-SALOMON V, BIALIK S, KIMCHI A. DAP-kinase and autophagy. Apoptosis. 2014;19(2):346-356. [73] GURKAR AU, CHU K, RAJ L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun. 2013;4:2189. [74] MAEJIMA Y, KYOI S, ZHAI P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478-1488. [75] WU H, XUE Y, ZHANG Y, et al. PTH1-34 promotes osteoblast formation through Beclin1-dependent autophagic activation. J Bone Miner Metab. 2021;39(4):572-582. [76] YANG X, JIANG T, WANG Y, et al. The role and mechanism of sirt1 in resveratrol-regulated osteoblast autophagy in osteoporosis rats. Sci Rep. 2019;9(1):18424. [77] BRACHMANN CB, SHERMAN JM, DEVINE SE, et al. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9(23):2888-2902. [78] TANG BL. Sirt1 and the mitochondria. Mol Cells. 2016;39(2):87-95. [79] WANG L, XU C, JOHANSEN T, et al. SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy. 2021;17(2):593-595. [80] NG F, TANG BL. Sirtuins’ modulation of autophagy. J Cell Physiol. 2013; 228(12):2262-2270. [81] LOUVET L, LETERME D, DELPLACE S, et al. Sirtuin 1 deficiency decreases bone mass and increases bone marrow adiposity in a mouse model of chronic energy deficiency. Bone. 2020;136:115361. [82] SHAO Z, DOU S, ZHU J, et al. Apelin-36 protects HT22 cells against oxygen-glucose deprivation/reperfusion-induced oxidative stress and mitochondrial dysfunction by promoting SIRT1-mediated PINK1/Parkin-dependent mitophagy. Neurotox Res. 2021;39(3):740-753. [83] DZULKO M, PONS M, HENKE A, et al. The PP2A subunit PR130 is a key regulator of cell development and oncogenic transformation. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188453. [84] MAZHAR S, LEONARD D, SOSA A, et al. Challenges and reinterpretation of antibody-based research on phosphorylation of Tyr307 on PP2Ac. Cell Rep. 2020;30(9):3164-3170.e3. [85] XU Y, WEI L, TANG S, et al. Regulation PP2Ac methylation ameliorating autophagy dysfunction caused by Mn is associated with mTORC1/ULK1 pathway. Food Chem Toxicol. 2021;156:112441. [86] HUSSAIN T, ZHAO D, SHAH SZA, et al. PP2Ac modulates AMPK-mediated induction of autophagy in mycobacterium bovis-infected macrophages. Int J Mol Sci. 2019;20(23):6030. [87] MENG L, LU C, WU B, et al. Taurine antagonizes macrophages M1 polarization by mitophagy-glycolysis switch blockage via dragging SAM-PP2Ac transmethylation. Front Immunol. 2021;12:648913. [88] OKAMURA H, YOSHIDA K, MORIMOTO H, et al. Role of protein phosphatase 2A in osteoblast differentiation and function. J Clin Med. 2017;6(3):23. [89] ONISHI M, YAMANO K, SATO M, et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40(3):e104705. [90] LOU G, PALIKARAS K, LAUTRUP S, et al. Mitophagy and neuroprotection. Trends Mol Med. 2020;26(1):8-20. [91] PICKRELL AM, YOULE RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257-273. [92] SINGLETON AB, FARRER MJ, BONIFATI V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord. 2013;28(1):14-23. [93] NARDIN A, SCHREPFER E, ZIVIANI E. Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson’s disease. Curr Neuropharmacol. 2016;14(3):250-259. [94] NGUYEN TN, PADMAN BS, LAZAROU M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26(10):733-744. [95] BINGOL B, SHENG M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210-222. [96] WANG B, ABRAHAM N, GAO G, et al. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl Neurodegener. 2016; 5:19. [97] ZHANG W, HOU W, CHEN M, et al. Upregulation of parkin accelerates osteoblastic differentiation of bone marrow-derived mesenchymal stem cells and bone regeneration by enhancing autophagy and β-catenin signaling. Front Cell Dev Biol. 2020;8:576104. [98] CHEN L, SHI X, WENG SJ, et al. Vitamin K2 can rescue the dexamethasone-induced downregulation of osteoblast autophagy and mitophagy thereby restoring osteoblast function in vitro and in vivo. Front Pharmacol. 2020; 11:1209. |
| [1] | Nong Fuxiang, Jiang Zhixiong, Li Yinghao, Xu Wencong, Shi Zhilan, Luo Hui, Zhang Qinglang, Zhong Shuang, Tang Meiwen. Bone cement augmented proximal femoral nail antirotation for type A3.3 intertrochanteric femoral fracturalysis [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(在线): 1-10. |
| [2] | Pan Zhongjie, Qin Zhihong, Zheng Tiejun, Ding Xiaofei, Liao Shijie. Targeting of non-coding RNAs in the pathogenesis of the osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1441-1447. |
| [3] | Cai Zhihao, Xie Zhaoyong. Femoral neck anteversion measurement assessment: how to establish a unified method and standard [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1448-1454. |
| [4] | Dang Yi, Du Chengyan, Yao Honglin, Yuan Nenghua, Cao Jin, Xiong Shan, Zhang Dingmei, Wang Xin. Hormonal osteonecrosis and oxidative stress [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1469-1476. |
| [5] | Wang Ji, Zhang Min, Yang Zhongya, Zhang Long. A review of physical activity intervention in type 2 diabetes mellitus with sarcopenia [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1272-1277. |
| [6] | Nie Chenchen, Su Kaiqi, Gao Jing, Fan Yongfu, Ruan Xiaodi, Yuan Jie, Duan Zhaoyuan, Feng Xiaodong. The regulatory role of circular RNAs in cerebral ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1286-1291. |
| [7] | Gao Yu, Han Jiahui, Ge Xin. Immunoinflammatory microenvironment after spinal cord ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1300-1305. |
| [8] | Zhao Lu, Zhao Yifei, Gao Da, Liu Yanfang, Fu Tingting, Xu Jiangyan. Expression of suppressor of Zeste 12 in kidney tissues of rats with diabetic nephropathy [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(8): 1179-1186. |
| [9] | Liu Xiaolin, Mu Xinyue, Ma Ziyu, Liu Shutai, Wang Wenlong, Han Xiaoqian, Dong Zhiheng. Effect of hydrogel-loaded simvastatin microspheres on osteoblast proliferation and differentiation [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 998-1003. |
| [10] | Xu Xingxing, Wen Chaoju, Meng Maohua, Wang Qinying, Chen Jingqiao, Dong Qiang. Carbon nanomaterials in oral implant [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1062-1070. |
| [11] | Li Cheng, Zheng Guoshuang, Kuai Xiandong, Yu Weiting. Alginate scaffold in articular cartilage repair [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1080-1088. |
| [12] | Chen Shisong, Liu Xiaohong, Xu Zhiyun. Current status and prospects of bioprosthetic heart valves [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1096-1102. |
| [13] | Shi Yehong, Wang Cheng, Chen Shijiu. Early thrombosis and prevention of small-diameter blood vessel prosthesis [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1110-1116. |
| [14] | Tang Haotian, Liao Rongdong, Tian Jing. Application and design of piezoelectric materials for bone defect repair [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(7): 1117-1125. |
| [15] | Liu Wentao, Feng Xingchao, Yang Yi, Bai Shengbin. Effect of M2 macrophage-derived exosomes on osteogenic differentiation of bone marrow mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(6): 840-845. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||