中国组织工程研究 ›› 2023, Vol. 27 ›› Issue (9): 1469-1476.doi: 10.12307/2022.958
• 骨与关节综述 bone and joint review • 上一篇
激素型骨坏死与氧化应激
党 祎1,杜成砚2,姚红林2,袁能华2,曹 金1,熊 山1,张顶梅1,王 信1
- 遵义医科大学附属医院,1骨科, 2手术室,贵州省遵义市 563003
-
收稿日期:2021-12-30接受日期:2022-01-30出版日期:2023-03-28发布日期:2022-07-02 -
通讯作者:王信,博士,主任医师,博士生导师,遵义医科大学附属医院骨科,贵州省遵义市 563003 张顶梅,博士,遵义医科大学附属医院骨科,贵州省遵义市 563003 -
作者简介:党祎,男,1997年生,河南省南阳市人,汉族,遵义医科大学在读硕士,主要从事骨缺损早期愈合的基础研究。 杜成砚,1983年生,汉族,贵州省遵义市人。 -
基金资助:国家自然科学基金(31960209,31760266),项目负责人:王信;贵州省科技厅基础研究计划(黔科合基础[2020]1Y093),项目负责人:王信;贵州省卫生健康委科学技术基金(gzwkj2022-108),项目负责人:张顶梅;遵义医科大学研究生科研基金(ZYK63),项目负责人:曹金;遵义医科大学研究生科研基金(ZYK57),项目负责人:熊山
Hormonal osteonecrosis and oxidative stress
Dang Yi1, Du Chengyan2, Yao Honglin2, Yuan Nenghua2, Cao Jin1, Xiong Shan1, Zhang Dingmei1, Wang Xin1
- 1Department of Orthopedics, 2Operating Room, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou Province, China
-
Received:2021-12-30Accepted:2022-01-30Online:2023-03-28Published:2022-07-02 -
Contact:Wang Xin, MD, Chief physician, Doctoral supervisor, Department of Orthopedics, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou Province, China Zhang Dingmei, MD, Department of Orthopedics, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou Province, China -
About author:Dang Yi, Master candidate, Department of Orthopedics, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou Province, China Du Chengyan, Operating Room, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou Province, China Dang Yi and Du Chengyan contributed equally to this article. -
Supported by:National Natural Science Foundation of China, No. 31960209, 31760266 (to WX); Basic Research Program of Guizhou Science and Technology Department, No. Guizhou Science and Technology Foundation [2020]1Y093 (to WX); Science and Technology Fund of Guizhou Provincial Health Commission, No. gzwkj2022-108 (to ZDM); Graduate Research Fund of Zunyi Medical University, No. ZYK63 (to CJ); Graduate Research Fund of Zunyi Medical University, No. ZYK57 (to XS))
摘要:
文题释义:
氧化应激:即氧化作用与非氧化作用失衡,其主要是线粒体损伤并释放大量的活性氧,并造成内源性抗氧化防御机制之间的失调。过量的活性氧会导致细胞和血管功能障碍,最终导致不可逆的细胞损伤和凋亡。
激素型骨坏死:过度使用糖皮质激素会导致患者发生非创伤性骨坏死,其主要表现为骨循环中断或受损,引起局部骨组织内细胞死亡,继而骨结构发生改变,从而导致患者关节功能障碍。
背景:糖皮质激素诱导骨坏死的确切机制尚不清楚,氧化应激是重要的机制之一。
目的:综述目前已知相关文献,总结激素型骨坏死与氧化应激之间的关系,了解氧化应激在激素型骨坏死中的发病机制。
方法:检索中国知网、万方、维普、中国生物医学文献数据库和PubMed数据库中与激素型骨坏死和氧化应激的研究进展相关的文献,并通过阅读摘要进行初筛,最终共纳入96篇文章进行综述分析。
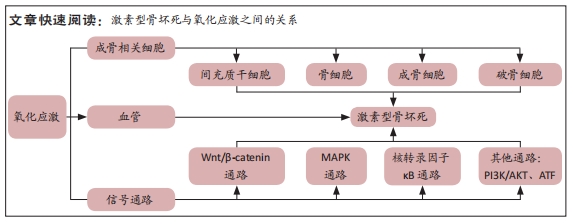
结果与结论:①活性氧将间充质干细胞、骨细胞、成骨细胞、破骨细胞及血管之间所形成的信号交流网络紧密地联系在一起,抑制其成骨和血管生成,从而在激素型股骨头坏死中发挥着重要作用;②文章分别阐释了活性氧在骨代谢中的作用情况,其主要通过Wnt/β-catenin、MAPK、核转录因子κB和PI3K/AKT等信号通路调控骨代谢中的细胞增殖、凋亡及分化,从而介导糖皮质激素所导致的骨形成和吸收失衡;③由于目前激素型骨坏死的机制比较复杂,其具体机制过程尚未明确;另外由于目前技术限制,抗氧化策略并未对疾病产生非常显著的影响;④未来应基于现有的相关机制,进一步了解氧化应激在激素型股骨头坏死骨组织损伤中的机制研究;并进一步分析活性氧参与调控的不同信号通路,从而为临床治疗骨坏死提供治疗靶点的研究方向。
https://orcid.org/0000-0001-7325-0674 (党祎);https://orcid.org/0000-0001-5015-3396 (王信)
中国组织工程研究杂志出版内容重点:人工关节;骨植入物;脊柱;骨折;内固定;数字化骨科;组织工程
中图分类号:
引用本文
党 祎, 杜成砚, 姚红林, 袁能华, 曹 金, 熊 山, 张顶梅, 王 信. 激素型骨坏死与氧化应激[J]. 中国组织工程研究, 2023, 27(9): 1469-1476.
Dang Yi, Du Chengyan, Yao Honglin, Yuan Nenghua, Cao Jin, Xiong Shan, Zhang Dingmei, Wang Xin. Hormonal osteonecrosis and oxidative stress[J]. Chinese Journal of Tissue Engineering Research, 2023, 27(9): 1469-1476.
糖皮质激素诱导的骨缺血坏死是一种代谢性疾病,它是由于使用糖皮质激素类药物,从而导致骨头血液供应受损、骨细胞死亡,进而导致骨结构性变化和关节功能障碍等[3]。该病的发展可能会对骨造成不可逆性的损伤,导致骨和关节功能障碍,严重影响患者的生活质量,若治疗不及时,可能会发展为骨关节炎等严重疾病,因而早期诊断、早期治疗非常重要。因此,阐明其发病机制对于该病的防治具有重要的指导意义。已有研究表明,在糖皮质激素诱导股骨头坏死的过程中,发现骨代谢失衡与氧化应激有关[13]。抑制糖皮质激素诱导的氧化损伤,可以有效减少成骨细胞和骨细胞凋亡,保护骨组织[14]。从目前来讲,激素型骨缺血坏死的机制假说过程尚不明确,研究者们认为这是多种因素共同作用的结果,如血管假说、细胞凋亡和氧化应激等等[3]。
2.2 氧化应激 氧化应激被定义为活性氧产生和内源性抗氧化防御机制之间的失调(即氧化与非氧化作用失衡)[15],主要表现为活性氧水平增加。活性氧包括多种活性分子和自由基,如超氧阴离子、过氧化氢和羟基自由基,在有氧呼吸过程中,其电子传递链上产生的分子可以影响细胞信号和稳态等生物功能[16]。活性氧主要来源于线粒体,具有调节细胞稳态功能和介导细胞内信号等生理作用,是调节细胞增殖、存活、代谢、凋亡、分化和迁移的必需物质[17]。过量的活性氧破坏细胞内氧化还原平衡,导致线粒体DNA突变,使细胞发生不可逆损伤,从而导致疾病发生[6,18]。烟酰胺腺嘌呤二核苷酸磷酸氧化酶是一种产生活性氧的酶系统。在骨骼中,烟酰胺腺嘌呤二核苷酸磷酸氧化酶来源的活性氧诱导成骨细胞或骨细胞的线粒体功能障碍和凋亡,从而造成成骨细胞和破骨细胞的骨稳态失衡,最终导致骨组织弱化,并可能引发骨缺血坏死[17,19-21]。氧化应激与骨稳态密切相关,活性氧过量产生导致大鼠成骨细胞凋亡,并抑制其增殖和成骨功能,最终引发骨病[22],而使用活性氧清除剂N-乙酰半胱氨酸清除体内过量活性氧可逆转氧化应激诱导的成骨细胞凋亡[23-24]。
2.3 氧化应激在激素型骨缺血坏死中的作用
2.3.1 氧化应激与骨代谢 骨组织由多种细胞构成,主要包括促进骨形成的成骨细胞、促进骨吸收的破骨细胞以及成熟骨组织内的骨细胞等[25]。骨组织具有强大的自我修复能力,在正常情况下,成骨细胞和破骨细胞协同作用,共同维持骨形成和骨吸收平衡[5]。间充质干细胞可分化为成骨细胞,从而调节成骨细胞和破骨细胞之间的平衡。活性氧可以直接影响间充质干细胞、骨细胞、成骨细胞和破骨细胞的活力和功能[26-27],见图3。
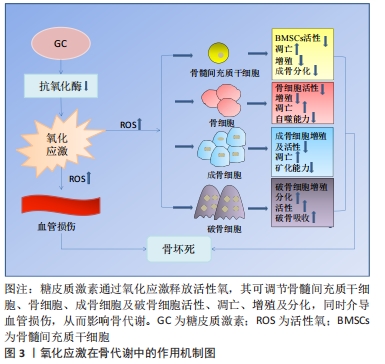
氧化应激对骨髓间充质干细胞的影响:骨髓间充质干细胞是一种多能性细胞,具有自我更新和多种分化潜能,它可以分化成软骨细胞、成骨细胞、脂肪细胞等多种细胞类型[28]。由于成骨细胞和脂肪细胞分化之间相互联系,骨髓间充质干细胞分化为一种谱系后逐渐丧失向另一种谱系分化的潜能[29]。
研究发现氧化应激诱导骨髓间充质干细胞凋亡,而使用青蒿素降低活性氧水平后,其活性得到改善[30]。此外,活性氧水平增加可诱导骨髓间充质干细胞线粒体功能障碍,促进细胞凋亡[31]。抑制活性氧生成还可促进骨髓间充质干细胞细胞周期,从而促进细胞增殖[32]。另有研究者使用白藜芦醇抑制内源性活性氧生成,发现其可促进衰老骨髓间充质干细胞的成骨分化[33]。因此,活性氧可通过诱导氧化应激,损害骨髓间充质干细胞抗氧化防御系统,对其活性、凋亡、增殖以及成骨分化产生不利影响。
氧化应激对骨细胞的影响:骨细胞是骨组织最丰富的细胞,在骨重建中发挥多种功能,包括调节成骨和破骨细胞吸收[34]。骨坏死本质上就是骨细胞的死亡,高剂量的糖皮质激素治疗增加了骨细胞凋亡[35-36],这是骨缺血坏死的重要原因[21]。骨细胞作为一种力学传感器,在骨损伤和修复中发挥作用,其丢失会破坏骨细胞-小管网络从而破坏骨结构,糖皮质激素可以通过改变骨细胞陷窝周围骨组织的弹性模量增加骨脆性,从而影响骨细胞功能[37]。在骨重塑过程中,细胞内氧化还原状态发生变化,氧化应激抑制骨细胞活性[17]。糖皮质激素处理骨细胞可显著提高细胞活性氧水平,清除活性氧则可阻止骨细胞凋亡[26]。WANG等[38]使用特立帕肽处理骨细胞,发现其可降低糖皮质激素增加的细胞活性氧水平,促进细胞增殖。由此可见,特立帕肽通过抑制活性氧在成骨过程中发挥着重要作用。此外,KAR等[39]发现,活性氧增加导致骨细胞自噬活性下降,自噬激活为细胞抗氧化应激提供了保护,抑制自噬增加了活性氧诱导的骨细胞死亡。这些研究说明氧化应激可影响骨细胞活性、增殖及自噬等,从而介导糖皮质激素诱导的骨细胞凋亡。
氧化应激对成骨细胞的影响:成骨细胞来源于间充质干细胞,对于维持骨量和骨骼发育是不可或缺的,可分化为骨细胞并调控骨基质矿化[40-41]。在激素型骨缺血坏死中,成骨细胞凋亡且功能减退,导致骨丢失[42]。暴露在地塞米松环境下,成骨细胞内活性氧水平增加,通过抑制碱性磷酸酶、成骨特异性转录因子(Runt-related transcription factor 2,RUNX2)、Ⅰ型胶原、骨形态发生蛋白2、骨连接素和骨钙素等多种成骨标志物的表达从而导致细胞功能障碍,引起细胞死亡[18,43]。活性氧清除剂通过消除体内活性氧的产生,逆转氧化应激诱导的成骨细胞凋亡[23-24],增加成骨细胞活性[44],并诱导前成骨细胞的分化[45]。PENG等[46]的研究发现,地塞米松引起活性氧水平增加促进了成骨细胞凋亡,使用N-乙酰半胱氨酸消除过量活性氧显著降低细胞凋亡比例。过多生成的活性氧损伤成骨细胞的脂质和蛋白质合成,不仅导致细胞损伤和凋亡[41],还会使得细胞周期阻滞且增殖能力下降[47]。此外,成骨细胞黏附有助于骨基质的合成和矿化[48],活性氧过度产生会损害这一过程[49]。综上,活性氧不仅可抑制成骨细胞的增殖与分化、降低成骨活性以及增加细胞凋亡,还可影响成骨矿化能力,最终导致成骨功能障碍、骨形成降低。
氧化应激对破骨细胞的影响:破骨细胞来自造血祖细胞,是造血前体细胞衍生的多核细胞,它具有骨吸收功能。形成成熟的破骨细胞需依赖于核因子κB受体活化因子配体(receptor activator of nuclear factor kappa B ligand,RANKL)[50]。破骨细胞的分化和活性可通过RANKL与核因子κB受体激活因子(receptor activator of nuclear factor Kappa B,RANK)相互作用来激活,介导骨吸收[51]。破骨细胞过度活跃是糖皮质激素诱导股骨头坏死的重要原因[52]。活性氧在破骨细胞的形成和骨吸收中起着至关重要的作用,其可以控制破骨细胞的分化成熟[53]。此外,活性氧可促进破骨细胞增殖[38]。研究发现,过氧化氢可显著增加破骨细胞数量和活性[54],而抗氧化剂则降低破骨细胞的分化和活性[17]。活性氧是骨丢失的关键要素,不管哪种活性氧清除剂都可以通过去除活性氧抑制破骨吸收。HAN等[55]使用三白草酮减少活性氧的产生,发现其显著降低与破骨相关基因如碳酸酐酶Ⅱ、基质金属蛋白酶9和抗酒石酸酸性磷酸酶表达水平,从而抑制破骨吸收。另有报道,百里醌可通过抑制活性氧生成,从而下调破骨相关基因抗酒石酸酸性磷酸酶、树突状细胞特异性跨膜蛋白等表达,抑制破骨细胞形成[56]。这些研究证实了活性氧可促进破骨细胞增殖、分化并增强其活性,从而促进破骨吸收,这可能是糖皮质激素诱导骨坏死的重要机制之一。因此,通过降低活性氧水平抑制破骨细胞增殖分化,最终起到修复坏死区骨组织的作用。
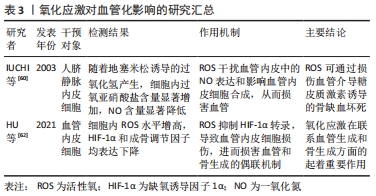
2.3.2 氧化应激对血管的影响 糖皮质激素诱导骨缺血坏死的两大基础是细胞凋亡和缺血[57]。血管作为营养提供者,它在骨组织中至关重要,并在骨重塑中发挥着重要作用。研究表明,血管异常导致血栓形成和栓塞,进而导致骨缺血坏死的发生[58]。另有研究表明,股骨头缺血坏死患者随着骨内压逐渐增高,窦道受压,静脉血管淤滞,最终引起动脉阻塞,导致骨缺血性坏死[59]。为了防止糖皮质激素诱导的骨缺血坏死,保护血管就显得非常重要。
活性氧不仅对骨代谢有调控作用,而且对血管有重要的调控作用。糖皮质激素可通过产生大量的活性氧直接作用于内皮细胞,对其造成损伤,导致骨毛细血管数量减少,凝血-纤溶系统紊乱,血栓形成,严重降低骨小梁的血供[59-60]。一氧化氮是内皮细胞衍生的舒张因子,是内皮依赖性血管舒张反应的重要血管活性物质。活性氧干扰血管内皮中的一氧化氮表达和影响血管内皮细胞合成,从而损害血管[60]。此外,缺氧诱导因子1α在血管生成中发挥着重要作用,活性氧可抑制其表达[61]。HU等[62]在高糖环境下培养骨特异性血管内皮细胞,发现细胞内活性氧水平增高,缺氧诱导因子1α和成骨调节因子均表达下降,而在使用抗氧化剂后细胞功能障碍得以改善,这表明活性氧诱导的血管内皮细胞损伤可造成血管数目减少和功能障碍,进而损害血管和骨生成的偶联机制,从而导致骨形成受损。由此可知,活性氧可通过损伤血管介导糖皮质激素诱导的骨缺血坏死。血管生成可为坏死区骨组织修复提供养料,从而在修复骨坏死过程中发挥重要作用。但由于氧化应激在联系血管生成和骨生成方面的具体机制有待进一步阐明,因此,后续研究可将其作为激素性股骨头坏死的研究重点。
2.4 活性氧通过信号通路影响骨坏死 根据相关实验研究表明,目前氧化应激在激素性骨坏死骨组织修复过程中有多条信号通路参与,其中主要以干预Wnt/β-catenin、MAPK、核转录因子κB等通路来促进骨吸收、抑制骨形成,从而降低骨量,见图4。
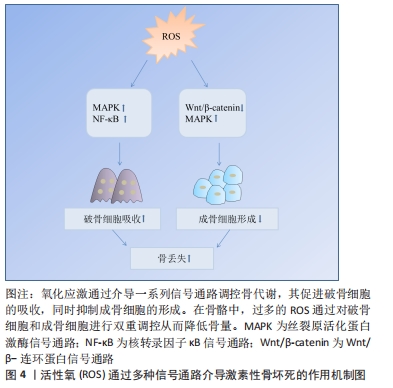
2.4.1 Wnt/β-catenin信号通路 Wnt/β-catenin信号通路又称为典型的Wnt信号通路,是一个进化保守的信号轴,参与细胞的分化、增殖和凋亡等生理过程[63],该信号通路参与调节成骨细胞和骨细胞[64-65]。在Wnt配体缺失的情况下,胞质β-catenin被磷酸化,磷酸化的β-catenin被蛋白酶体系统进一步泛素化并迅速降解,以防止在细胞质积累;Wnt配体存在时,细胞质中蓄积β-catenin,可易位到细胞核,与相关转录因子结合启动成骨细胞分化相关基因转录[63,66-69]。升高的活性氧通过转移β-catenin池,抑制成骨分化并导致骨细胞和成骨细胞凋亡增加[20,70]。此外,活性氧还可通过增强叉头盒O表达,减弱小鼠成骨细胞祖细胞中β-catenin转录,抑制Wnt/β-catenin信号通路,从而降低成骨细胞增殖,减弱骨形成[71]。这些研究提示,活性氧可通过直接或间接作用于Wnt/β-catenin信号通路,从而参与糖皮质激素诱导的骨缺血坏死。由此可见,活性氧通过介导Wnt/β-catenin信号通路,无论在细胞增殖、凋亡方面,还是结合骨组织工程应用上,对于治疗激素性股骨头坏死都具有十分重要的研究价值。
2.4.2 MAPK信号通路 MAPKs是丝氨酸-苏氨酸蛋白激酶的一个家族,其家族成员包括细胞外信号调节激酶(extracellular signal-regulated kinase,ERKs)、c-JunN端激酶(c-Jun N terminal kinase,JNKs)和p38 MAPKs,参与信号转导、细胞分化、增殖、凋亡和免疫应答等[72-73]。ERK和p38 MAPKs可磷酸化成骨细胞分化的主要调控因子RUNX2,从而促进成骨分化;而JNK在破骨细胞的凋亡、形成和分化中发挥着重要作用[74-75]。此外,MAPK/EPK信号通路参与间充质干细胞的增殖、存活及分化过程,激活该信号通路可促进细胞增殖,提高细胞活力[76],若该通路失活可阻断成骨细胞分化,促进间充质干细胞分化为脂肪细胞[77]。活性氧可以抑制MARK磷酸酶,从而提高MARK表达[41]。在原代成骨细胞和骨髓间充质干细胞中,过氧化氢通过刺激ERK1/2磷酸化,导致成骨细胞分化受损[78]。氧化应激还可通过激活JNK,p38和ERK1/2介导RANKL刺激的破骨细胞分化[79]。由此可见,氧化应激通过调节MAPK信号通路调节骨稳态,从而参与糖皮质激素诱导的骨坏死过程。
2.4.3 核转录因子κB信号通路 核转录因子κB由一系列转录因子组成,包括p65/RelA,RelB,cRel,p50和p52,在炎症、免疫、细胞增殖、分化和生存中发挥关键作用[80]。有研究发现,核转录因子κB信号通路的激活与糖皮质激素相关股骨头坏死密切相关[81]。核转录因子κB是一种重要的氧化还原敏感转录因子,可调节破骨细胞和成骨细胞,在骨形成和骨吸收中发挥重要作用[82],抑制核转录因子κB可导致骨形成增加和骨吸收减少[83]。RANKL是核转录因子κB信号通路的重要调控因子[84],其可通过诱导核转录因子κB活化,刺激破骨细胞标记基因的表达,从而促进破骨细胞前体细胞分化形成成熟的破骨细胞[83,85]。RANKL刺激破骨细胞产生活性氧,用抗氧化剂预处理破骨细胞可降低RANKL诱导的核转录因子κB的激活[86],表明RANKL通过活性氧诱导核转录因子κB活化促进破骨形成。活性氧还可通过激活核转录因子κB,促进破骨细胞形成和活性[87]。有研究发现,作为一种强抗氧化剂,硫辛酸可以通过抑制活性氧产生和核转录因子κB的活化,从而抑制RANKL诱导骨髓前体细胞中破骨细胞生成[88]。核转录因子κB活性主要由核因子κB抑制蛋白调控,活性氧通过核因子κB抑制蛋白α(NF-kappa-B inhibitor alpha,IKBα)磷酸化来激活核转录因子κB[89]。已有研究表明,谷胱甘肽过氧化物酶过表达导致活性氧水平下降,通过抑制IKB-α磷酸化,破坏磷酸化IKB-α的特异性蛋白水解,从而导致核转录因子κB激活[90]。综上所述,文章认为活性氧在核转录因子κB信号交流网络中,可通过传递RANKL来调节破骨细胞的生物活动,介导破骨生成,最终可能导致骨缺血坏死等疾病发生。
2.4.4 其他信号通路 除上述通路外,活性氧还可通过磷脂酰肌醇-4,5-二磷酸3-激酶/蛋白激酶B(Phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B,PI3K/AKT)、激活转录因子(Activating transcription factor,ATF)等信号通路参与激素型骨缺血坏死的过程[20,91]。例如,PI3K/AKT信号通路参与细胞增殖、分化、侵袭和凋亡等过程[92]。PI3K的激活导致AKT的磷酸化和激活,AKT通过增强抗凋亡蛋白的表达和抑制促凋亡蛋白的活性来促进细胞存活[93]。激活PI3K/AKT信号通路是大鼠成骨细胞增殖分化的必要条件[94],通过使用具有较强抗氧化活性的四甲基吡嗪预处理,FANG等[95]发现其促进骨髓间充质干细胞中p-AKT的表达。这证明PI3K/AKT信号通路参与了四甲基吡嗪对抗氧化应激诱导的骨髓间充质干细胞凋亡。CHEN等[96]也指出,ATF-3在刺激活性氧产生后,促进了小鼠成骨细胞中RANKL基因表达增加。由此可知,无论是体内还是体外实验,皆证实了在氧化应激的条件下,许多关键通路参与骨细胞的增殖、分化和凋亡。每种途径都代表一种潜在的治疗方法,可通过这些信号通路进一步研究设计防止骨丢失的抗氧化策略。
2.5 氧化应激与骨形成
2.5.1 活性氧对骨形成的影响 活性氧不仅可以通过诱导氧化应激直接作用于骨髓间充质干细胞、骨细胞和成骨细胞,损害其抗氧化防御系统,还可以通过介导Wnt/β-catenin、MAPK和PI3K/AKT信号通路抑制骨形成:①活性氧增加可诱导MAPK磷酸化,导致成骨细胞功能受损;②活性氧可通过减弱成骨细胞中β-catenin转录,抑制Wnt/β-catenin信号通路,从而减弱骨形成;③PI3K的激活导致AKT磷酸化和激活,AKT通过增强抗凋亡蛋白的表达和抑制促凋亡蛋白的活性来促进细胞存活。活性氧可通过介导PI3K/AKT信号通路参与骨缺血坏死的发生。氧化应激对骨形成影响汇总表,见表1。

2.5.2 活性氧与骨吸收的关系 活性氧可通过促进破骨细胞的形成和增加破骨细胞活性来调节骨吸收:①RANKL与RANK相互作用可刺激破骨细胞的分化及活性,过量的活性氧可通过上调RANKL的表达促进骨吸收;②RANKL是核转录因子κB信号通路的重要调控因子,其可通过活性氧诱导核转录因子κB活化,刺激破骨细胞标记基因的表达,从而促进破骨细胞分化成熟;③氧化应激通过调节MAPK信号通路调节骨代谢,其可通过激活JNK、p38 MAPKs和ERK1/2介导RANKL刺激的破骨细胞分化;④活性氧水平增高导致氧化还原失衡,破骨细胞形成及活性增加;使用抗氧化药物下调活性氧水平,可抑制破骨细胞形成,减少骨吸收。氧化应激对骨吸收影响汇总表,见表2。

2.5.3 氧化应激在联系血管生成和骨生成方面发挥着重要作用 ①血管内皮细胞通过释放多种舒缩因子如一氧化氮来调节血管张力,而活性氧可干扰血管内皮中一氧化氮表达,影响血管内皮合成,进而损害血管;②缺氧诱导因子1α促进血管生成,而活性氧可抑制其表达,损伤血管内皮细胞,进而损害血管和骨生成的偶联机制,从而损伤骨形成。氧化应激对血管化影响汇总表,见表3。
| [1] GUERADO E, CASO E. The physiopathology of avascular necrosis of the femoral head: an update. Injury. 2016;47 Suppl 6:S16-S26. [2] KERACHIAN MA, SEGUIN C, HARVEY EJ. Glucocorticoids in osteonecrosis of the femoral head: a new understanding of the mechanisms of action. J Steroid Biochem Mol Biol. 2009;114(3-5):121-128. [3] WANG A, REN M, WANG J. The pathogenesis of steroid-induced osteonecrosis of the femoral head: a systematic review of the literature. Gene. 2018;671:103-109. [4] CHANG C, GREENSPAN A, GERSHWIN ME. The pathogenesis, diagnosis and clinical manifestations of steroid-induced osteonecrosis. J Autoimmun. 2020;110:102460. [5] BAI SC, XU Q, LI H, et al. NADPH oxidase isoforms are involved in glucocorticoid-induced preosteoblast apoptosis. Oxid Med Cell Longev. 2019;2019:9192413. [6] MIURA Y. Oxidative stress, radiation-adaptive responses, and aging. J Radiat Res. 2004;45(3):357-372. [7] GESSI S, MERIGHI S, BOREA PA. Glucocorticoid’s pharmacology: past, present and future. Curr Pharm Des. 2010;16(32):3540-3553. [8] WEINSTEIN RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin North Am. 2012;41(3):595-611. [9] FENG ZH, ZHENG WH, TANG Q, et al. Fludarabine inhibits STAT1-mediated up-regulation of caspase-3 expression in dexamethasone-induced osteoblasts apoptosis and slows the progression of steroid-induced avascular necrosis of the femoral head in rats. Apoptosis. 2017;22(8):1001-1012. [10] ZALAVRAS C, SHAH S, BIRNBAUM MJ, et al. Role of apoptosis in glucocorticoid-induced osteoporosis and osteonecrosis. Crit Rev Eukaryot Gene Expr. 2003;13(2-4):221-235. [11] ZHAO D, ZHANG F, WANG BJ, et al. Guidelines for clinical diagnosis and treatment of osteonecrosis of the femoral head in adults (2019 version). J Orthop Translat. 2020;21:100-110. [12] PETEK D, HANNOUCHE D, SUVA D. Osteonecrosis of the femoral head: pathophysiology and current concepts of treatment. EFORT Open Rev. 2019;4(3):85-97. [13] ZHU SY, ZHUANG JS, WU Q, et al. Advanced oxidation protein products induce pre-osteoblast apoptosis through a nicotinamide adenine dinucleotide phosphate oxidase-dependent, mitogen-activated protein kinases-mediated intrinsic apoptosis pathway. Aging Cell. 2018;17(4): e12764. [14] FAN ZQ, BAI SC, XU Q, et al. Oxidative stress induced osteocyte apoptosis in steroid-induced femoral head necrosis. Orthop Surg. 2021; 13(7):2145-2152. [15] VAN DER POL A, VAN GILST WH, VOORS AA, et al. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail. 2019; 21(4):425-435. [16] AGIDIGBI TS, KIM C. Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated osteoclast diseases. Int J Mol Sci. 2019;20(14):3576. [17] DOMAZETOVIC V, MARCUCCI G, IANTOMASI T, et al. Oxidative stress in bone remodeling: role of antioxidants. Clin Cases Miner Bone Metab. 2017;14(2):209-216. [18] KUBO Y, DRESCHER W, FRAGOULIS A, et al. Adverse effects of oxidative stress on bone and vasculature in corticosteroid-associated osteonecrosis: potential role of nuclear factor erythroid 2-related factor 2 in cytoprotection. antioxid redox signal. 2021;35(5):357-376. [19] WERNER SL, SHARMA R, WOODRUFF KA, et al. CSF-1 in osteocytes inhibits nox4-mediated oxidative stress and promotes normal bone homeostasis. JBMR Plus. 2020;4(7):e10080. [20] WAUQUIER F, LEOTOING L, COXAM V, et al. Oxidative stress in bone remodelling and disease. Trends Mol Med. 2009;15(10):468-477. [21] CALDER JD, BUTTERY L, REVELL PA, et al. Apoptosis-a significant cause of bone cell death in osteonecrosis of the femoral head. J Bone Joint Surg Br. 2004;86(8):1209-1213. [22] UENO T, YAMADA M, IGARASHI Y, et al. N-acetyl cysteine protects osteoblastic function from oxidative stress. J Biomed Mater Res A. 2011;99(4):523-531. [23] FENG H, LI ZY, DU J, et al. Dual function of peroxiredoxin I in lipopolysaccharide-induced osteoblast apoptosis via reactive oxygen species and the apoptosis signal-regulating kinase 1 signaling pathway. Cell Death Discov. 2018;4:47. [24] DU J, FENG W, SUN J, et al. Ovariectomy upregulated the expression of Peroxiredoxin 1 &5 in osteoblasts of mice. Sci Rep. 2016;6:35995. [25] NOLLET M, SANTUCCI-DARMANIN S, BREUIL V, et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy. 2014;10(11):1965-1977. [26] YINJ, HAN L, CONG W. Alpinumisoflavone rescues glucocorticoid-induced apoptosis of osteocytes via suppressing Nox2-dependent ROS generation. Pharmacol Rep. 2018;70(2):270-276. [27] CHEN XJ, SHEN YS, HE MC, et al. Polydatin promotes the osteogenic differentiation of human bone mesenchymal stem cells by activating the BMP2-Wnt/beta-catenin signaling pathway. Biomed Pharmacother. 2019;112:108746. [28] RUIZ M, COSENZA S, MAUMUS M, et al. Therapeutic application of mesenchymal stem cells in osteoarthritis. Expert Opin Biol Ther. 2016; 16(1):33-42. [29] MURUGANANDAN S, ROMAN AA, SINAL CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci. 2009;66(2):236-253. [30] FANG J, ZHAO X, LI S, et al. Protective mechanism of artemisinin on rat bone marrow-derived mesenchymal stem cells against apoptosis induced by hydrogen peroxide via activation of c-Raf-Erk1/2-p90(rsk)-CREB pathway. Stem Cell Res Ther. 2019;10(1):312. [31] WEI H, LI ZW, HU SS, et al. Apoptosis of mesenchymal stem cells induced by hydrogen peroxide concerns both endoplasmic reticulum stress and mitochondrial death pathway through regulation of caspases, p38 and JNK. J Cell Biochem. 2010;111(4):967-978. [32] SUN LY, PANG CY, LI DK, et al. Antioxidants cause rapid expansion of human adipose-derived mesenchymal stem cells via CDK and CDK inhibitor regulation. J Biomed Sci. 2013;20(1):53. [33] ZHOU T, YAN YR, ZHAO CC, et al. Resveratrol improves osteogenic differentiation of senescent bone mesenchymal stem cells through inhibiting endogenous reactive oxygen species production via AMPK activation. Redox Rep. 2019;24(1):62-69. [34] CAPULLI M, PAONE R, RUCCI N. Osteoblast and osteocyte: games without frontiers. Arch Biochem Biophys. 2014;561:3-12. [35] COMPTON JT, LEE FY. A review of osteocyte function and the emerging importance of sclerostin. J Bone Joint Surg Am. 2014;96(19):1659-1668. [36] JILKA RL, NOBLE B, WEINSTEIN RS. Osteocyte apoptosis. Bone. 2013; 54(2):264-271. [37] CANALIS E, MAZZIOTTI G, GIUSTINA A, et al. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18(10):1319-1328. [38] WANG T, HAN C, TIAN P, et al. Role of teriparatide in glucocorticoid-induced osteoporosis through regulating cellular reactive oxygen species. Orthop Surg. 2018;10(2):152-159. [39] KAR R, RIQUELME MA, HUA R, et al. Glucocorticoid-induced autophagy protects osteocytes against oxidative stress through activation of MAPK/ERK signaling. JBMR Plus. 2019;3(4):e10077. [40] NAKASHIMA K,DE CROMBRUGGHE B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003; 19(8):458-466. [41] TAO H, GE G, LIANG XL, et al. ROS signaling cascades: dual regulations for osteoclast and osteoblast. Acta Biochim Biophys Sin (Shanghai). 2020;52(10):1055-1062. [42] CHEN F, ZHANG L, OUYANG YP, et al. Glucocorticoid induced osteoblast apoptosis by increasing E4BP4 expression via up-regulation of Bim. Calcif Tissue Int. 2014;94(6):640-647. [43] NIE Z, DENG S, ZHANG L, et al. Crocin protects against dexamethasoneinduced osteoblast apoptosis by inhibiting the ROS/Ca2+ mediated mitochondrial pathway. Mol Med Rep. 2019;20(1):401-408. [44] XU G, LI X, ZHU ZY, et al. Iron overload induces apoptosis and cytoprotective autophagy regulated by ROS generation in Mc3t3-E1 cells. Biol Trace Elem Res. 2021;199(10):3781-3792. [45] PINNA A, BAGHBADERANI MT, HERNANDEZ VV, et al. Nanoceria provides antioxidant and osteogenic properties to mesoporous silica nanoparticles for osteoporosis treatment. Acta Biomater. 2021;122: 365-376. [46] PENG P, NIE ZG, SUN F, et al. Glucocorticoids induce femoral head necrosis in rats through the ROS/JNK/c-Jun pathway. FEBS Open Biol. 2021;11(1):312-321. [47] WANG Z, YANG XY, YANG SY, et al. Sodium fluoride suppress proliferation and induce apoptosis through decreased insulin-like growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts. Arch Toxicol. 2011;85(11):1407-1417. [48] FERNANDES GV, CAVAGIS AD, FERREIRA CV, et al. Osteoblast adhesion dynamics: a possible role for ROS and LMW-PTP. J Cell Biochem. 2014; 115(6):1063-1069. [49] ZHANG Y, YANG JH. Activation of the PI3K/Akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J Cell Biochem. 2013;114(11): 2595-2602. [50] GINGERY A, BRADLEY E, SHAW A, et al. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89(1):165-179. [51] HENRIKSEN K, NEUTZSKY-WULFF AV, BONEWALD LF, et al. Local communication on and within bone controls bone remodeling. Bone. 2009;44(6):1026-1033. [52] CHEN K, LIU YH, HE JB, et al. Steroid-induced osteonecrosis of the femoral head reveals enhanced reactive oxygen species and hyperactive osteoclasts. Int J Biol Sci. 2020;16(11):1888-1900. [53] SITHOLE C, PIETERSE C, HOWARD K, et al. GPR120 inhibits rankl-induced osteoclast formation and resorption by attenuating reactive oxygen species production in RAW264.7 Murine Macrophages. Int J Mol Sci. 2021;22(19):10544. [54] BAEK KH, OH KW, LEE WY, et al. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcif Tissue Int. 2010;87(3):226-235. [55] HAN KY, YANG D, CHANG EJ, et al. Inhibition of osteoclast differentiation and bone resorption by sauchinone. Biochem Pharmacol. 2007;74(6): 911-923. [56] AN J, HAO DJ, ZHANG Q, et al. Natural products for treatment of bone erosive diseases: The effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int Immunopharmacol. 2016; 36:118-131. [57] ZHANG Q, LV J, JIN L. Role of coagulopathy in glucocorticoid-induced osteonecrosis of the femoral head. J Int Med Res. 2018;46(6):2141-2148. [58] JONES LC, MONT MA, LE TB, et al. Procoagulants and osteonecrosis. J Rheumatol. 2003;30(4):783-791. [59] Kerachian MA, HARVEY EJ, COURNOYER D, et al. Avascular necrosis of the femoral head: vascular hypotheses. Endothelium. 2006;13(4):237-244. [60] IUCHI T, AKAIKE M, MITSUI T, et al. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res. 2003;92(1):81-87. [61] XIAO HJ, GU ZY, WANG GX, et al. The possible mechanisms underlying the impairment of HIF-1α pathway signaling in hyperglycemia and the beneficial effects of certain therapies. Int J Med Sci. 2013;10(10):1412-1421. [62] HU XF, XIANG G, WANG TJ, et al. Impairment of type H vessels by NOX2-mediated endothelial oxidative stress: critical mechanisms and therapeutic targets for bone fragility in streptozotocin-induced type 1 diabetic mice. Theranostics. 2021;11(8):3796-3812. [63] ZHANG Y, WANG X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J Hematol Oncol. 2020;13(1):165. [64] BONEWALD LF, JOHNSON ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42(4):606-615. [65] KARNER CM, LONG F. Wnt signaling and cellular metabolism in osteoblasts. Cell Mol Life Sci. 2017;74(9):1649-1657. [66] HE S, TANG S. WNT/beta-catenin signaling in the development of liver cancers. Biomed Pharmacother. 2020;132:110851. [67] CLEVERS H, NUSSE R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192-1205. [68] PAI SG, CARNEIRO BA, MOTA JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10(1): 101. [69] MAEDA K, TAKAHASHI N, KOBAYASHI Y. Roles of Wnt signals in bone resorption during physiological and pathological states. J Mol Med (Berl). 2013;91(1):15-23. [70] MANOLAGAS SC, ALMEIDA M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21(11): 2605-2614. [71] IYER S, AMBROGINI E, BARTELL SM, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123(8): 3409-3419. [72] XU F, XU JX, XIONG X, et al. Salidroside inhibits MAPK, NF-kappaB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep. 2019;24(1):70-74. [73] CALLAWAY DA, JIANG JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab. 2015;33(4):359-370. [74] IKEDA F, MATSUBARA T, TSURUKAI T, et al. JNK/c-Jun signaling mediates an anti-apoptotic effect of RANKL in osteoclasts. J Bone Miner Res. 2008;23(6):907-914. [75] DAVID JP, SABAPATHY K, HOFFMANN O, et al. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115(Pt 22):4317-4325. [76] LIANG JQ, LU F, GAN B, et al. Low-dose tubacin promotes BMSCs proliferation and morphological changes through the ERK pathway. Am J Transl Res. 2019;11(3):1446-1459. [77] MIYAOKA Y, TNANKA M, NAIKI T, et al. Oncostatin M inhibits adipogenesis through the RAS/ERK and STAT5 signaling pathways. J Biol Chem. 2006;281(49):37913-37920. [78] BAI XC, LU D, BAI J, et al. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem Biophys Res Commun. 2004; 314(1):197-207. [79] LEE NK, CHOI YG, BAIK JY, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106(3):852-859. [80] OECKINGHAUS A, GHOSH S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4): a000034. [81] PEI J, FAN LH, NAN K, et al. Excessive activation of TLR4/NF-kappaB interactively suppresses the canonical wnt/beta-catenin pathway and induces SANFH in SD rats. Sci Rep. 2017;7(1):11928. [82] JIMI E, AOKI K, SAITO H, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10(6):617-624. [83] KIM K, KIM JH, KIM I, et al. TRIM38 regulates NF-kappaB activation through TAB2 degradation in osteoclast and osteoblast differentiation. Bone. 2018;113:17-28. [84] LI J, YANG MY, LU CY, et al. Tuna bone powder alleviates glucocorticoid-induced osteoporosis via coregulation of the NF-kappab and Wnt/beta-catenin signaling pathways and modulation of gut microbiota composition and metabolism. Mol Nutr Food Res. 2020;64(5):e1900861. [85] KIM JH, KIM N. Signaling pathways in osteoclast differentiation. Chonnam Med J. 2016;52(1):12-17. [86] HA H, KWAK HB, LEE SW, et al. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res. 2004;301(2):119-127. [87] LEPETSOS P, PAPAVASSILIOU KA, PAPAVASSILIOU AG. Redox and NF-kappaB signaling in osteoarthritis. Free Radic Biol Med. 2019;132:90-100. [88] KIM HJ, CHANG EJ, KIM HM, et al. Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumor necrosis factor-alpha. Free Radic Biol Med. 2006;40(9):1483-1493. [89] MORGAN MJ, LIU ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103-115. [90] KRETZ-REMY C, MEHLEN P, MIRAULT ME, et al. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J Cell Biol. 1996; 133(5):1083-1093. [91] ZHAN J, YAN ZJ, ZHAO MY, et al. Allicin inhibits osteoblast apoptosis and steroid-induced necrosis of femoral head progression by activating the PI3K/AKT pathway. Food Funct. 2020;11(9):7830-7841. [92] MA P, GU B, XIONG W, et al. Glimepiride promotes osteogenic differentiation in rat osteoblasts via the PI3K/Akt/eNOS pathway in a high glucose microenvironment. PLoS One. 2014;9(11):e112243. [93] DOWNWARD J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15(2):177-182. [94] MA P, GU B, MA JL, et al. Glimepiride induces proliferation and differentiation of rat osteoblasts via the PI3-kinase/Akt pathway. Metabolism. 2010;59(3):359-366. [95] FANG Y, CHU LS, LI L, et al. Tetramethylpyrazine protects bone marrow-derived mesenchymal stem cells against hydrogen peroxide-induced apoptosis through PI3K/Akt and ERK1/2 pathways. Biol Pharm Bull. 2017;40(12):2146-2152. [96] CHEN JR, SHANKAR K, NAGARAJAN S, et al. Protective effects of estradiol on ethanol-induced bone loss involve inhibition of reactive oxygen species generation in osteoblasts and downstream activation of the extracellular signal-regulated kinase/signal transducer and activator of transcription 3/receptor activator of nuclear factor-kappaB ligand signaling cascade. J Pharmacol Exp Ther. 2008;324(1):50-59. |
| [1] | 方兴艳, 田侦丽, 赵哲仪, 文平, 谢婷婷. 亚砷酸钠对人脐静脉内皮细胞损伤及鞘氨醇激酶1/1-磷酸鞘氨醇信号轴的影响[J]. 中国组织工程研究, 2023, 27(在线): 1-7. |
| [2] | 唐辉宇, 侯 彪, 夏晓丹, 向 伟, 谢松林. 机械牵张应力对兔肢体截骨后动脉血管的影响[J]. 中国组织工程研究, 2023, 27(9): 1422-1426. |
| [3] | 潘钟杰, 秦志鸿, 郑铁军, 丁晓飞, 廖世杰. 股骨头坏死发病机制中非编码RNA的靶标性[J]. 中国组织工程研究, 2023, 27(9): 1441-1447. |
| [4] | 蔡志浩, 谢召勇. 股骨颈前倾角测量评估:如何建立统一的方法和标准[J]. 中国组织工程研究, 2023, 27(9): 1448-1454. |
| [5] | 陶 新, 徐 逸, 宋志文, 刘锦波. Hippo信号通路参与脊髓损伤的调控[J]. 中国组织工程研究, 2023, 27(4): 619-625. |
| [6] | 李世浩, 李 奇, 李 震, 张媛媛, 刘苗苗, 欧阳懿, 徐卫国. 前交叉韧带损伤与重建术后患者的足底压力及步态分析[J]. 中国组织工程研究, 2023, 27(4): 626-631. |
| [7] | 程明光, 张超宇, 庄康乐, 阮 鹏, 左 逸, 周正春, 孔 祥, 葛建军, 程光存. 体外构建Stanford A型主动脉夹层3D三维动态模拟图及个体化组织工程血管[J]. 中国组织工程研究, 2023, 27(3): 335-338. |
| [8] | 李 睿, 刘 珍, 郭子歌, 卢瑞杰, 王 晨. 纯钛表面载阿司匹林微球与聚多巴胺复合涂层促进成骨分化[J]. 中国组织工程研究, 2023, 27(3): 374-379. |
| [9] | 周 洁, 裴锡波, 万乾炳. 不对称伤口敷料的研究进展与生物应用[J]. 中国组织工程研究, 2023, 27(3): 434-440. |
| [10] | 陈镜桥, 李 英, 孟茂花, 徐星星, 王勤英, 王 欢, 陆 婧, 舒佳玉, 董 强. 富血小板纤维蛋白在口腔医学研究中的相关进展[J]. 中国组织工程研究, 2023, 27(3): 441-446. |
| [11] | 韩 涛, 郝建强, 李文波, 石 杰, 高秋明. 抗生素骨水泥治疗骨关节感染的优势与问题[J]. 中国组织工程研究, 2023, 27(3): 470-477. |
| [12] | 骆 帝, 梁学振, 阎博昭, 李嘉程, 许 波, 李 刚. 补肾活血胶囊对激素性股骨头坏死大鼠骨破坏的修复[J]. 中国组织工程研究, 2023, 27(2): 184-191. |
| [13] | 谢平金, 罗 臻, 卢启贵, 郭艳幸, 陈群群, 李飞龙. 川芎嗪和过表达miR-20b-5p干预早期膝骨关节炎模型大鼠滑膜、 软骨和软骨下骨血管新生的组织学变化[J]. 中国组织工程研究, 2023, 27(2): 237-245. |
| [14] | 赵 峰, 樊少卿, 程晓燕, 李小娜, 李长生, 马浩杰. miR-132-3p靶向调节Ptch1减轻慢性压迫性神经损伤小鼠神经性疼痛[J]. 中国组织工程研究, 2023, 27(2): 230-236. |
| [15] | 牛梓晗, 余 扬, 艾 江, 卜盼盼, 李文博, 苏日耶·热合曼, 马少林. 沙棘总黄酮干预兔耳增生性瘢痕组织块的消退[J]. 中国组织工程研究, 2023, 27(2): 258-263. |
骨坏死又称缺血性骨坏死,是临床上的一种常见疾病,它主要是由于骨内血液循环障碍导致局部骨细胞死亡、骨小梁坏死,继而骨结构发生改变[1],其分为创伤性和非创伤性。非创伤性骨坏死主要是由药物、乙醇和凝血异常等因素引起,其中糖皮质激素的使用是其发生的常见原因[2]。糖皮质激素因具有广泛的药理作用而在临床上被广泛使用,如用作内分泌不足的长期替代疗法,治疗严重感染或炎症等,疗效显著,但长期大剂量使用可造成一系列严重的不良反应,股骨头缺血性坏死便是其中较常见的一种[3]。糖皮质激素所致的骨缺血坏死发生机制尚不明确,可能与细胞凋亡、凝血功能异常、血管系统紊乱、氧化应激和内皮细胞功能障碍等相关[4]。
已有研究表明,地塞米松(一种合成型糖皮质激素)可通过诱导活性氧介导氧化损伤,从而影响骨内细胞和血管的活力及功能[5]。氧化应激的核心是线粒体受损,受损的线粒体释放大量活性氧,破坏细胞内氧化还原平衡[6],从而调控细胞命运。氧化应激作为细胞内重要的死亡调控机制之一,深入研究其与激素型骨缺血坏死间的关系可以为激素型骨缺血坏死的防治靶点提供理论基础。
因此,文章不仅回顾了氧化应激对间充质干细胞、成骨细胞、骨细胞、破骨细胞及血管的影响,还详细综述了活性氧通过调控信号通路来影响成骨的机制,以进一步了解骨坏死,希望能对抗氧化策略在治疗激素型骨坏死的基础、临床及骨组织工程应用方面的进一步研究提供一些帮助。
中国组织工程研究杂志出版内容重点:人工关节;骨植入物;脊柱;骨折;内固定;数字化骨科;组织工程
1.1.1 检索人及检索时间 第一作者在2021年10月进行检索。
1.1.2 检索文献时限 检索1996年1月至2021年12月PubMed数据库及1997年7月至2021年12月中国知网数据库相关文献。
1.1.3 检索数据库 中国知网、万方、维普、中国生物医学文献数据库和PubMed数据库。
1.1.4 检索词 英文检索词为“Glucocorticoid,Osteonecrosis,Oxidative stress,Bone metabolism,Osteocyte,Osteoblast,Osteoclast,Bone marrow mesenchymal stem cells,The blood vessels,Signal pathway”。中文检索词为“糖皮质激素、骨坏死、氧化应激、骨代谢、骨细胞、成骨细胞、破骨细胞、骨髓间充质干细胞、血管、信号通路”。
1.1.5 检索文献类型 检索文献类型为研究原著和综述。
1.1.6 手工检索情况 无。
1.1.7 检索策略 中英文数据库检索策略,见图1。
1.1.8 检索文献量 初步检索到中文文献556篇,英文文献4 807篇。
1.2 入组标准
1.2.1 纳入标准 ①文章内容与活性氧对激素型骨坏死、间充质干细胞、骨细胞、成骨细胞、破骨细胞、血管及相关信号通路密切相关;②同一领域选择新近文献或在权威杂志上发表的文章。
1.2.2 排除标准 筛除重复性研究及与研究目的无关的文献。
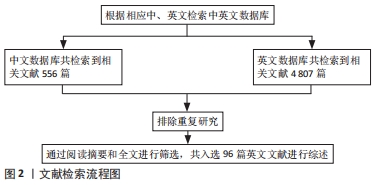
1.3 数据提取和文献质量评估 通过阅读标题和摘要初筛,排除与文章目的不符、年代久远及重复性研究,共入选英文文献96篇。认真阅读文献内容,总结相关有效信息并严谨撰写文章。文献检索流程图,见图2。
3.2 作者综述区别于他人他篇的特点 作者通过整理近年来氧化应激在激素型骨坏死的相关研究发现,活性氧通过将相关细胞因子转移至靶细胞,将骨髓间充质干细胞、骨细胞、成骨细胞、破骨细胞及血管之间所形成的信号交流网络紧密地联系在一起,抑制成骨分化和血管生成,从而介导糖皮质激素诱导的骨坏死,造成骨缺损和丢失。而且总结目前的相关机制研究发现,活性氧主要通过Wnt/β-catenin、MAPK、核转录因子κB和PI3K/AKT等信号通路调节骨代谢中细胞增殖、凋亡以及分化,从而促进破骨吸收及抑制成骨分化。针对目前抗氧化治疗骨坏死的研究现状,以期能为日后相关治疗靶点研究提供一些思考与参考。
3.3 综述的局限性 由于目前激素型骨坏死的机制比较复杂,其具体机制过程尚未明确。目前相关性研究多集中于单种信号通路异常,未能在文章中系统总结出信号通路机制方面的具体情况。另外由于目前技术限制,抗氧化策略并未完全成熟,相关信号通路作为治疗靶点的研发机制仍有待进一步开发研究。
3.4 综述的重要意义 活性氧可通过影响骨髓间充质干细胞、骨细胞、成骨细胞、破骨细胞和血管的功能,导致细胞凋亡和血液循环功能障碍,骨质平衡遭到破坏,机械支撑结构受损,骨结构发生改变,最终导致糖皮质激素诱导的骨缺血坏死发生。就目前研究状况而言,关于氧化应激在激素型骨缺血坏死发生发展中作用机制的研究虽然取得很大的进步,但由于骨系统代谢活跃,受到多种生长因子和信号通路调控,在一些疾病中,抗氧化策略并未对疾病起到非常积极的影响。因此,通过阐明氧化应激与各种细胞因子及信号通路的相互作用,从而保持骨形成和骨吸收之间的平衡,有望成为临床上预防和治疗骨缺血坏死的一种新的策略思想。
3.5 课题专家组对未来的建议 基于现有的相关机制,开展抗氧化在激素型骨坏死骨组织修复中的机制研究;通过进一步深入研究氧化应激与激素型骨坏死相关信号通路的联系,从而为临床应用提供一定的前提与基础。
中国组织工程研究杂志出版内容重点:人工关节;骨植入物;脊柱;骨折;内固定;数字化骨科;组织工程
文题释义:
氧化应激:即氧化作用与非氧化作用失衡,其主要是线粒体损伤并释放大量的活性氧,并造成内源性抗氧化防御机制之间的失调。过量的活性氧会导致细胞和血管功能障碍,最终导致不可逆的细胞损伤和凋亡。
激素型骨坏死:过度使用糖皮质激素会导致患者发生非创伤性骨坏死,其主要表现为骨循环中断或受损,引起局部骨组织内细胞死亡,继而骨结构发生改变,从而导致患者关节功能障碍。
氧化应激的特征是活性氧水平增加,破坏细胞内氧化还原平衡。
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||