Chinese Journal of Tissue Engineering Research ›› 2023, Vol. 27 ›› Issue (35): 5707-5713.doi: 10.12307/2023.599
Previous Articles Next Articles
Regulation of cardiovascular diseases by histone deacetylation modification
Han Weiyu, Chen Yuanxing, Huang Youyang, Liu Weiwei, Zhao Yongchao, Zhao Ranzun
- Department of Cardiovascular Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi 563000, Guizhou Province, China
-
Received:2022-10-08Accepted:2022-11-25Online:2023-12-18Published:2023-06-05 -
Contact:Zhao Ranzun, MD, Chief physician, Professor, Doctoral supervisor, Department of Cardiovascular Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi 563000, Guizhou Province, China -
About author:Han Weiyu, Master candidate, Department of Cardiovascular Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi 563000, Guizhou Province, China -
Supported by:National Natural Science Foundation of China, No. 81960066 (to ZRZ); Science and Technology Planning Project of Zunyi, Nos. HZ(2022)341 (to ZRZ) and HZ(2022)366 (to ZYC); Basic Research Program of Guizhou Science and Technology Department, Nos. ZK[2022] general 671 (to ZYC) and ZK[2022] general 653 (to LWW); Science and Technology Foundation of Guizhou Provincial Health Commission, No. gzwkj2021-103 (to LWW)
CLC Number:
Cite this article
Han Weiyu, Chen Yuanxing, Huang Youyang, Liu Weiwei, Zhao Yongchao, Zhao Ranzun. Regulation of cardiovascular diseases by histone deacetylation modification[J]. Chinese Journal of Tissue Engineering Research, 2023, 27(35): 5707-5713.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks

2.1 组蛋白去乙酰化修饰概述 组蛋白修饰是表观遗传学修饰的一种,也是转录激活或抑制标记。1884年A.KOSSEL首次从细胞核中鉴定出组蛋白的存在。随后又发现组蛋白中存在大量修饰氨基酸,直至1963年D.M.P.PHILLIPS首次发现组蛋白中存在乙酰化修饰的氨基酸,开辟了组蛋白乙酰化修饰的新方向。此后随着研究的深入,也逐步发现了组蛋白去乙酰化相关的酶分子家族以及相应的抑制剂。随着表观遗传学的深入,多种疾病模型中也证实了组蛋白去乙酰化修饰对各种疾病的影响及其作用机制,突出了组蛋白去乙酰化修饰对人类疾病的治疗前景,随后的研究也证明了组蛋白去乙酰化修饰对多种疾病和细胞的重要性,见图3。"


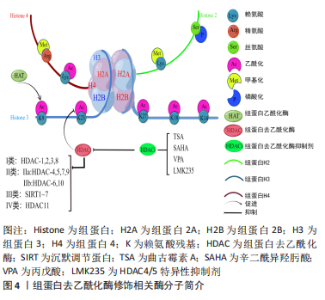
组蛋白是由核心组蛋白H2A,H2B,H3,H4各2个分子组成的八聚体[10],是真核生物染色质的基本单位(核小体)的主要组成成分,在核小体的核心,组蛋白组成一个球状结构域和一个氨基末端,氨基末端伸出核小体外,可发生甲基化、乙酰化、磷酸化、泛素化等修饰[11],其修饰异常可导致心血管疾病的发生。核心和接头组蛋白的序列变体的有序和局部掺入,尤其是通过组蛋白大量不同的翻译后修饰,这些翻译后修饰标记单个核小体或核小体片段,影响组蛋白与核小体之间的缠绕方式,进而影响染色质调控基因表达的功能,从而潜在地控制基础DNA的生物学功能[12]。 组蛋白乙酰化是一种特殊蛋白的修饰,一般认为组蛋白乙酰化修饰多数发生在组蛋白H3或H4的N端特定的赖氨酸残基上[13],有乙酰化和去乙酰化两方面,分别受组蛋白乙酰化酶(histone acetyltransferase,HAT)和组蛋白去乙酰化酶(histone deacetylase,HDAC)控制,组蛋白赖氨酸残基上的乙酰化修饰能中和残基上的正电荷,抑制组蛋白与DNA正负电荷的相互作用,进而破坏组蛋白与DNA的结合,导致染色质结构疏松,使转录因子以及修饰激活因子更容易接近DNA,影响DNA的转录与表达[14],乙酰化修饰的程度与状态决定基因是否表达。被乙酰化修饰的赖氨酸主要发生在H3,H4的赖氨酸(K)残基上(K9(H3K9),H3K14,H3K18,H3K23和H3K27)[15],以及其他不常见的H2A和H2B上,见表1。而去乙酰化则发挥着类似相反的作用,组蛋白去乙酰化酶包括18个成员,根据酵母细胞转录因子同源程度将组蛋白去乙酰化酶分为Ⅰ、Ⅱ、Ⅲ、Ⅳ类[16]。Ⅰ类有组蛋白去乙酰化酶1,2,3,8,在所有的细胞类型中均表达,含有核定位信号的序列,但无核输出信号序列,其主要作用是影响细胞的增殖活性[17];Ⅱ类分为Ⅱa、Ⅱb 类,其中Ⅱa 包括组蛋白去乙酰化酶4,5,7,9,Ⅱb 类包括 组蛋白去乙酰化酶 6,10[18],Ⅱ类酶含有核定位信号序列以及核输出信号序列,因此能够在细胞浆和细胞核之间穿梭,其主要作用是参与细胞的分化;Ⅲ类组蛋白去乙酰化酶是需依赖烟酰胺腺嘌呤二核苷酸激活的SIRT家族,主要是SIRT 1-7,其作用不同于Ⅰ、Ⅱ和Ⅳ类[19],主要是调控单核细胞的凋亡;Ⅳ类为 Zn+ 依赖的 组蛋白去乙酰化酶11[20]。组蛋白去乙酰化酶抑制剂(histone deaceylase inhibitors,HDACi)可以有效抑制组蛋白去乙酰化,组蛋白乙酰化程度对基因表达、染色体结构、表观调控等方面起到重要调节作用,从而调控基因的表达。而组蛋白去乙酰化酶抑制剂主要有代表Ⅰ类的曲古霉素A(trichostatin A,TSA)[21]、辛二酰异羟肟酸(suberoylanilide hydroxamic acid,SAHA/vorinostat)[22]、丙戊酸(valproic acid,VPA)以及组蛋白去乙酰化酶4,5特异性抑制剂LMK235等[23-24],见图4。"

"


2.2 组蛋白去乙酰化修饰与心血管疾病 去乙酰化修饰是一类翻译后修饰过程,通过表观遗传学调控蛋白质的多种化学性质,如赖氨酸残基上的可逆组蛋白乙酰化调控DNA和组蛋白之间的相互作用,介导染色质重塑和基因转录。 非组蛋白去乙酰化使细胞功能复杂化,而关键线粒体酶的乙酰化调节生物能量代谢[25]。功能蛋白的乙酰化和去乙酰化在胚胎发育[26]、组织细胞的成熟[27]、心肌细胞分化[28]、心脏重塑和各种心血管疾病(包括肥胖、糖尿病、心脏代谢疾病、缺血-再灌注损伤、心脏重塑、高血压和心律失常)的微妙的稳态调节中至关重要[29],如表2所示。组蛋白乙酰转移酶和组蛋白去乙酰化酶是主要调控赖氨酸乙酰化水平的必要酶,为心血管疾病的治疗干预提供了可能的药物靶点。"


2.2.1 组蛋白去乙酰化修饰与动脉粥样硬化 研究发现,组蛋白去乙酰化酶对细胞增殖、迁移和内皮及平滑肌细胞的凋亡有一定作用[50],因此有可能成为动脉粥样硬化新型治疗靶点。在动脉粥样硬化发生发展过程中,组蛋白修饰相关酶的表达水平显著差异变化[51-52],动脉粥样硬化发生的危险因素如脂多糖以及低密度脂蛋白,可通过调节组蛋白修饰来调节相关基因的表达,从而诱导内皮细胞功能障碍。研究已证明组蛋白去乙酰化酶1,2,3可在正常内皮细胞中表达,而在动脉粥样硬化斑块的内皮细胞中,其表达量及活性均明显降低。NAN等[30]在动脉粥样硬化小鼠模型以及氧化型低密度脂蛋白处理的人脐静脉内皮细胞中发现组蛋白去乙酰化酶1可被miR-410靶向Krüppel样因子5(Krüppel-like factor-5,KLF5),增加核因子?B抑制蛋白α(NF-?appa-B inhibitor alpha,IkBα)的表达,从而抑制动脉粥样硬化中核因子?B(nuclear factor kappa B,NF-?B)的表达,而在动脉粥样硬化小鼠中过表达组蛋白去乙酰化酶1可增加细胞活性,抑制凋亡和炎症反应,从而阻止动脉粥样硬化的发展。此外,在高脂饮食饲养的低密度脂蛋白基因敲除小鼠模型中,缺乏髓系组蛋白去乙酰化酶2可降低雄性小鼠动脉粥样硬化39%,通过使得组蛋白3乙酰化增加,增加谷氧还蛋白1(glutaredoxin 1,Grx1)的表达,进而发挥动脉粥样硬化保护作用[31]。ZAMPETAKI等[32]研究发现,内皮细胞的完整性由组蛋白去乙酰化酶上调丝氨酸-酪氨酸激酶的活性来维持,在动脉血流紊乱的分支血管附近的组蛋白去乙酰化酶3基因上调,并且在载脂蛋白E敲除(ApoE-/-)小鼠敲减组蛋白去乙酰化酶3后会形成严重动脉粥样硬化病变。在此基础上,WANG等[33]进一步研究发现组蛋白去乙酰化酶3上调可通过上调miR-19b介导的过氧化物酶体增殖物活化受体γ(peroxisome proliferator-activated receptorgamma,PPARγ)的表达使得NF-?B/p65失活,从而阻止炎症抑制动脉粥样硬化的发生发展,为动脉粥样硬化的治疗提供了一个基本的治疗方案。同时,组蛋白去乙酰化酶11作为Ⅳ型组蛋白去乙酰化酶亚家族中的唯一成员,在动脉粥样硬化的发生发展中也发挥着重要作用,YAO等[34]发现高脂饮食喂养8周或12周的ApoE-/-小鼠的主动脉组蛋白去乙酰化酶11表达上调,并出现内皮细胞下垂现象,而进一步研究发现,组蛋白去乙酰化酶11与Ets基因家族相关基因(ETS-related gene,ERG)形成络合物并降低Ets基因家族相关基因的乙酰化水平,促进细胞焦亡通路的发生,该研究表明,组蛋白去乙酰化酶11的调控可能成为动脉粥样硬化治疗策略的潜在靶点。 综上所述,组蛋白去乙酰化修饰在动脉粥样硬化的发生发展中起着重要作用,主要通过影响动脉粥样硬化斑块的内皮细胞的功能障碍,介导内皮细胞在炎症、自噬、凋亡、焦亡以及细胞完整性等通路中的去乙酰化修饰作用,进而影响动脉粥样硬化过程中脂质斑块的形成与发展。组蛋白去乙酰化修饰调控动脉粥样硬化的进展有望成为抗动脉粥样硬化治疗的新靶点。 2.2.2 组蛋白去乙酰化修饰与心力衰竭 心力衰竭是一种反复住院、病死率高的进展性疾病,是遗传易感性和环境因素相互作用的结果,也是大多数心脏疾病的终末发展阶段。病理性心肌细胞肥大也称之为心力衰竭,因此探索新的治疗方法预防或者逆转病理性心肌细胞肥大至关重要。随着转录后修饰的发现和深入研究,近些年来越来越多的研究证实组蛋白修饰参与了心肌肥大的发生发展过程,其与心肌细胞的病理性肥大、凋亡、心肌间质纤维化密切相关[53],清除心力衰竭细胞的细胞外基质,从而控制心肌细胞肥大的程度。研究发现,在人类、小鼠和猪衰竭的心脏中,Ⅲ类组蛋白去乙酰化酶沉默信息调节因子1(silence information regulation factor 1,SIRT1)的表达水平均显著降低,通过进一步探究发现SIRT1的下调增加了内质网钙离子ATP酶(sarcoplasmic reticulum calcium ion atpase,SERCA2a)的乙酰化水平,而当激活SIRT1的药理学活性增加SERCA2a的去乙酰化水平时,由心衰导致的心脏功能障碍得到明显的缓解[35],表明SIRT1可作为治疗心力衰竭的新靶标。各种数据表明,Ⅰ类和Ⅱ类组蛋白去乙酰化酶都参与调节心脏肥大的过程,但是它们发挥的作用却截然相反。Ⅰ类组蛋白去乙酰化酶2,3,8具有促心肌细胞肥大的作用,改善心脏功能,例如组蛋白去乙酰化酶8基因敲除或者组蛋白去乙酰化酶8选择性抑制可降低p38促分裂原活化的蛋白激酶(mitogen activated protein kinase,MAPK)的激活,从而抑制异丙肾上腺素诱导的心肌肥大和纤维化[36],表明组蛋白去乙酰化酶8可能通过调节p38 MAPK信号通路而成为治疗心肌肥大和纤维化的一个有前途的靶点。同样的,组蛋白去乙酰化酶3在心衰大鼠和肥大细胞中都高表达,组蛋白去乙酰化酶3通过去乙酰化修饰DNA甲基转移酶1(DNA-methyltransferase 1,DNMT1),抑制泛素化介导的降解,抑制非受体型蛋白质酪氨酸磷酸酶1(Src homology region 2 domain-containing phosphatase 1,SHP-1)的表达,进而导致心肌肥大[37]。在腹主动脉部分收缩诱导的大鼠心肌肥大模型中,组蛋白去乙酰化酶2同样起着促进心肌肥大的作用[38]。相反,Ⅱa类的组蛋白去乙酰化酶以不依赖酶活性的方式可通过抑制心肌特异性转录因子,如肌细胞增强因子2(myocyte enhancer factor 2,MEF2)[54]、GATA结合蛋白4和活化T细胞核因子等抑制心脏肥大[55]。许多研究证明Ⅱ类组蛋白去乙酰化酶尤其是Ⅱa类(组蛋白去乙酰化酶4,5,7,9)作为心肌肥大的负性调节剂,可通过结合DNA特定区域抑制心肌肥大的转录活动增强子2的活性,而抑制组蛋白去乙酰化酶4可改善心肌梗死诱导的心衰小鼠的心功能[39]。组蛋白去乙酰化酶5,9基因敲除小鼠会因压力负荷过大而发生心肌肥大[56-58],相应地,过表达组蛋白去乙酰化酶2则会阻止相同刺激导致的肥大反应。Ⅱa类组蛋白去乙酰化酶的抗肥大活性与其结合并随后抑制成年心肌细胞中的肌细胞增强因子2的能力有关[59],Ⅱa类组蛋白去乙酰化酶通过与细胞核中的肌细胞增强因子2结合,抑制组蛋白去乙酰化酶的活性,然而在蛋白激酶C以及蛋白激酶D等激酶存在的情况下,与肌细胞增强因子2结合的组蛋白去乙酰化酶被磷酸化而从细胞核转移至细胞质中,随后组蛋白去乙酰化酶磷酸化突变体显示出对PKC信号通路的抗性并阻断心肌细胞的肥大发展[56],对于大多数Ⅱa类组蛋白去乙酰化酶抑制心脏肥大的效果是由核浆-细胞浆在应激信号变化时介导组蛋白去乙酰化酶从细胞核穿梭到细胞质这一过程效果中来调控的,造成这种相反效果的原因可能与胞核和胞质组蛋白去乙酰化酶作用的靶点效应不同相关。 综上所述,组蛋白去乙酰化修饰在心力衰竭的发生发展中起着非常重要的作用,主要通过心肌细胞肥大、心肌间质的纤维化等方面进行调控。然而不同种类的组蛋白去乙酰化酶针对心肌肥大有着不同甚至相反的作用效应,导致这种差异的具体原因除了上述研究中发现的核浆-细胞浆应激信号变化原因导致的组蛋白去乙酰化酶出核之外,其他原因还有待进一步去探究。 2.2.3 组蛋白去乙酰化修饰与心肌梗死 近年来,组蛋白去乙酰化酶被证明可以控制心血管疾病的发展和恶化,包括心肌梗死以及梗死后的心肌缺血再灌注损伤[60]。WNT诱导的信号通路蛋白1(WNT-inducible secreted protein-1,Wisp-1)是一种母体细胞蛋白,可促进血管生成和分离的心肌细胞存活,在心肌梗死损伤后,WNT诱导的信号通路蛋白1转录受组蛋白去乙酰化酶调节,并可由组蛋白去乙酰化酶抑制剂辛二酰异羟肟酸修饰。据研究发现,在心肌梗死发生后7 d,相对于单独的心肌梗死患者,在遭受心肌梗死损伤并每天注射辛二酰异羟肟酸的小鼠中,梗死边界区的WNT诱导的信号通路蛋白1升高了3倍,而Wisp通过刺激心肌细胞产生血管内皮生长因子α(vascular endothelial growth factor α,VEGF-α),促进血管生成,进而减少心肌梗死后的梗死面积[40]。由于缺乏足够的氧合和潜在的血运重建,心肌梗死导致心肌细胞的丢失,演变为心肌纤维化的产生,组蛋白去乙酰化酶抑制剂辛二酰异羟肟酸可通过促进双特异性磷酸酶4(Dual specificity phosphatase 4,DUSP4)的表达抑制转化生长因子β1(transforming growth factor β1,TGF-β1)诱导的P38磷酸化而发挥抗心肌梗死后诱导心脏纤维化的发生与发展[22]。超声显示辛二酰异羟肟酸治疗可改善心肌缺血再灌注损伤后心肌收缩功能,辛二酰异羟肟酸对心脏的保护作用有赖于自噬泡的内吞,治疗后的心肌梗死边缘带发现大量自噬体形成。TIAN等[41]在大鼠心肌梗死不同时间点、发病时间和损伤后分别给予组蛋白乙酰化抑制剂丙戊酸治疗,结果显示丙戊酸治疗可能是通过叉头框蛋白M1(forked frame protein M1,Foxm1)途径,调控对细胞生存和抗炎相关的基因的表达,使得心肌梗死时氧化应激和细胞死亡显著降低,将急性心肌梗死后的梗死面积减少约50%,起到明显的心脏保护作用,长期治疗可显著改善心肌功能。同样地,WANG等[42]在小鼠上的实验也证明组蛋白去乙酰化酶抑制剂曲古霉素A可预防心肌梗死后的心脏重塑,并且可以逆转低氧诱导的自噬过程,使得心肌梗死细胞的数量减少40%。 组蛋白去乙酰化酶抑制剂已成为减少心肌缺血再灌注损伤,恢复心脏功能和减少梗塞面积的新型药物。抑制组蛋白去乙酰化酶,尤其是Ⅰ类组蛋白去乙酰化酶,对缺血再灌注后的心肌具有保护作用,例如Ⅰ类组蛋白去乙酰化酶选择性抑制剂莫西替诺坦可通过激活环磷酸腺苷反应元件结合蛋白(cAMP response element binding protein,CREB)和上调过氧化物酶体增殖物激活受体γ共激活因子1α (peroxisome proliferator activated receptor γ coactivator-1α,PGC-1α)来保护心肌缺血再灌注所致的损伤[43]。小鼠心肌细胞特异性过表达组蛋白去乙酰化酶4可增加心肌缺血再灌注损伤梗死面积,导致心肌中自噬蛋白微管相关蛋白1轻链3(autophagy-related proteins microtubule-associated protein 1 light chain 3,LC3)增加,而超氧化物歧化酶1(superoxyde dismutase 1,SOD-1)减少,组蛋白去乙酰化酶抑制剂的传递减弱了活性组蛋白去乙酰化酶4对心肌缺血再灌注损伤的有害作用[44]。HUANG等[45]试着进一步探究糖尿病患者心肌梗死死亡率和发病率较高是否也与翻译后组蛋白乙酰化修饰相关,于是采用非糖尿病和糖尿病小鼠骨髓内皮祖细胞培养液中的细胞外囊泡,观察其对体外永久性结扎左前降支和缺血/再灌注心肌缺血损伤的修复作用,结果发现糖尿病小鼠骨髓内皮祖细胞源细胞外囊泡通过降低组蛋白3赖氨酸9乙酰化的水平进而对心脏内皮细胞的血管生成、细胞存活和增殖产生影响,而使用组蛋白去乙酰化酶抑制剂丙戊酸治疗后可显著改善糖尿病小鼠骨髓内皮祖细胞介导的心肌梗死后心脏功能的修复。 综上所述,组蛋白去乙酰化修饰在缺血性心肌梗死以及梗死后的再灌注损伤中发挥重大作用,去乙酰化修饰反应主要是在缺血梗死部位周围新生血管的生成、缺血部位细胞的生存活性、周围炎症及自噬反应、梗死后心脏纤维化及心室重塑等生物学过程中发挥调控作用,企图通过组蛋白去乙酰化抑制剂等方法促进梗死以及再灌注损伤后的心肌与血管的修复,达到对心肌梗死的治疗效果。近几年的基础研究结果显示,组蛋白去乙酰化酶抑制剂在心肌梗死以及梗死后再灌注损伤过程中的损伤修复效果非常明显,有望进一步在临床中进行转化应用。 2.2.4 组蛋白去乙酰化修饰与高血压 已发现组蛋白修饰与高血压中血管功能的调节有关。先前有研究表明,组蛋白去乙酰化酶在高血压的发生发展中发挥着重要作用,其相应的抑制剂具有明显的抗高血压的作用,尤其是Ⅲ类组蛋白去乙酰化酶抑制剂。例如SIRT1的过表达在体内可降低血管紧张素Ⅱ引起的体内血管重构以及高血压,并减少活性氧的生成,抑制氧化应激和凋亡[46]。DIKALOVA等[47]在SIRT3敲除的转基因小鼠中及在血管紧张素Ⅱ和醋酸去氧皮质酮盐诱导的高血压模型中同样发现类似的现象,SIRT3的全面缺失导致线粒体超氧化物歧化酶2的过度乙酰化导致氧化应激,增加缺氧诱导因子1α(hypoxia-inducible factor 1-α,HIF-1α),刺激血管内皮肥厚,增加血管通透性和血管炎症,加速血管衰老和年龄依赖性高血压,同时SIRT3衰竭的临床相关性在原发性高血压患者的纵膈脂肪小动脉中也得到证实,显示血管SIRT3降低40%。GUO等[48]发现,在醋酸盐和血管紧张素Ⅱ诱导的两种独立的高血压小鼠中,内皮SIRT6的水平显著降低,而在内皮特异性SIRT6敲除后血压显著增高,加剧了实验性高血压患者的内皮功能障碍和心肾损伤,从机制上讲,SIRT6通过使组蛋白H3K9脱乙酰基来抑制人同源盒基因(NK3 homeobox 2,Nkx3.2)转录,从而诱导GATA结合蛋白5(GATA-binding protein 5,GATA5)介导的信号传导途径,从而防止了内皮损伤,因此SIRT6在体内过表达为醋酸盐以及血管紧张素诱导的高血压的治疗潜力提供了直接的证据。新型组蛋白去乙酰化酶抑制剂 CG200745可通过抑制醋酸去氧皮质酮引起高血压大鼠的心肌肥大和纤维化进而减轻高血压。YOON等[49]进一步研究发现,CG200745还可以通过抑制组蛋白去乙酰化酶/血管紧张素Ⅱ/血管收缩轴降低高脂血症鼠的血压、组蛋白去乙酰化酶活性,肾素和血管紧张素原以及血清血管紧张素Ⅱ水平,进而改善高脂血症诱发的高血压,为CG200745作为高脂血症诱发高血压提供了新的选择。 综上所述,组蛋白去乙酰化修饰在血管紧张素及醋酸盐诱导的高血压模型以及原发性高血压患者中发挥重要作用,尤其是Ⅲ类SIRT去乙酰化酶,大多数高血压模型中都表现为去乙酰化酶表达降低、去乙酰化程度降低,主要通过调控血管内皮细胞的通透性、内皮细胞的活性及损伤等过程,进而调控心脏收缩和舒张过程中血管壁的压力。因此,Ⅲ类去乙酰化酶SIRT可能是干预高血压发生发展危险因素以及治疗的特异性靶点,有望开发SIRT抑制剂进行高血压疾病的治疗,减缓高血压疾病的进展以及高血压导致的其他器官的损伤作用。"

| [1] 《中国心血管健康与疾病报告》编写组.《中国心血管健康与疾病报告2020》要点解读[J]. 中国心血管杂志,2021,26(3):209-218. [2] ZHAO D, LIU J, WANG M, et al. Epidemiology of cardiovascular disease in China: current features and implications. Nat Rev Cardiol. 2019;16(4):203-212. [3] PRASHER D, GREENWAY SC, SINGH RB. The impact of epigenetics on cardiovascular disease. Biochem Cell Biol. 2020;98(1):12-22. [4] LACAGNINA S. Epigenetics. Am J Lifestyle Med. 2019;13(2):165-169. [5] CAVALLI G, HEARD E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489-499. [6] LEE HT, OH S, RO DH, et al. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J Lipid Atheroscler. 2020;9(3): 419-434. [7] CHELLADURAI P, BOUCHERAT O, STENMARK K, et al. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br J Pharmacol. 2021;178(1):54-71. [8] FUNAMOTO M, SUNAGAWA Y, KATANASAKA Y, et al. Histone Acetylation Domains Are Differentially Induced during Development of Heart Failure in Dahl Salt-Sensitive Rats. Int J Mol Sci. 2021;22(4):1771. [9] LEI I, TIAN S, GAO W, et al. Acetyl-CoA production by specific metabolites promotes cardiac repair after myocardial infarction via histone acetylation. Elife. 2021;10:e60311. [10] SUDA M, IWAI K. Identification of suberimidate cross-linking sites of four histone sequences in H1-depleted chromatin. Histone arrangement in nucleosome core. J Biochem. 1979;86(6):1659-1670. [11] LIU Y, MIN J. Structure and function of histone methylation-binding proteins in plants. Biochem J. 2016;473(12):1663-1680. [12] TSE C, HANSEN JC. Hybrid trypsinized nucleosomal arrays: identification of multiple functional roles of the H2A/H2B and H3/H4 N-termini in chromatin fiber compaction. Biochemistry. 1997;36(38):11381-11388. [13] XU H, YE M, XIA A, et al. The Fng3 ING protein regulates H3 acetylation and H4 deacetylation by interacting with two distinct histone-modifying complexes. New Phytol. 2022;235(6):2350-2364. [14] KOUZARIDES T. Chromatin modifications and their function. Cell. 2007;128(4): 693-705. [15] BHAUMIK S R, SMITH E, SHILATIFARD A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14(11): 1008-1016. [16] ROPERO S, ESTELLER M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007;1(1):19-25. [17] SANCHEZ-FERNANDEZ C, LORDA-DIEZ C I, DUARTE-OLIVENZA C, et al. Histone Epigenetic Signatures in Embryonic Limb Interdigital Cells Fated to Die. Cells. 2021;10(4):911. [18] SCHLüTER A, AKSAN B, FIORAVANTI R, et al. Histone Deacetylases Contribute to Excitotoxicity-Triggered Degeneration of Retinal Ganglion Cells In Vivo. Mol Neurobiol. 2019;56(12):8018-8034. [19] HE Q, CHEN K, YE R, et al. Associations of sirtuins with clinicopathological variables and prognosis in human ovarian cancer. Oncol Lett. 2020;19(4):3278-3288. [20] NúñEZ-ÁLVAREZ Y, SUELVES M. HDAC11: a multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2022;289(10):2771-2792. [21] KONG Y, REN W, FANG H, et al. Histone Deacetylase Inhibitors (HDACi) Promote KLF5 Ubiquitination and Degradation in Basal-like Breast Cancer. Int J Biol Sci. 2022;18(5):2104-2115. [22] WANG K, TANG R, WANG S, et al. SAHA could inhibit TGF-β1/p38 pathway in MI-induced cardiac fibrosis through DUSP4 overexpression. Heart Vessels. 2022; 37(1):152-160. [23] RUESS DA, PROBST M, MARJANOVIC G, et al. HDACi Valproic Acid (VPA) and Suberoylanilide Hydroxamic Acid (SAHA) Delay but Fail to Protect against Warm Hepatic Ischemia-Reperfusion Injury. PLoS One. 2016;11(8):e0161233. [24] LIU Z, CHEN T, HAN Q, et al. HDAC inhibitor LMK‑235 promotes the odontoblast differentiation of dental pulp cells. Mol Med Rep. 2018;17(1):1445-1452. [25] BALPARDA M, ELSäSSER M, BADIA M B, et al. Acetylation of conserved lysines fine-tunes mitochondrial malate dehydrogenase activity in land plants. Plant J. 2022;109(1):92-111. [26] SARAIVA N Z, OLIVEIRA C S, ALMEIDA N, et al. Epigenetic modifiers during in vitro maturation as a strategy to increase oocyte competence in bovine. Theriogenology. 2022;187:95-101. [27] TAO L, YU H V, LLAMAS J, et al. Enhancer decommissioning imposes an epigenetic barrier to sensory hair cell regeneration. Dev Cell. 2021;56(17):2471-2485.e5. [28] WANG D, LIU C, LI Z, et al. Regulation of Histone Acetylation on Expression Profiles of Potassium Channels During Cardiomyocyte Differentiation From Mouse Embryonic Stem Cells. J Cell Biochem. 2017;118(12):4460-4467. [29] PONS D, DE VRIES FR, VAN DEN ELSEN PJ, et al. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur Heart J. 2009;30(3):266-277. [30] NAN S, WANG Y, XU C, et al. Interfering microRNA-410 attenuates atherosclerosis via the HDAC1/KLF5/IKBα/NF-κB axis. Mol Ther Nucleic Acids. 2021;24:646-657. [31] WANG L, AHN Y J, ASMIS R. Inhibition of myeloid HDAC2 upregulates glutaredoxin 1 expression, improves protein thiol redox state and protects against high-calorie diet-induced monocyte dysfunction and atherosclerosis. Atherosclerosis. 2021;328:23-32. [32] ZAMPETAKI A, ZENG L, MARGARITI A, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121(1):132-142. [33] WANG J, XU X, LI P, et al. HDAC3 protects against atherosclerosis through inhibition of inflammation via the microRNA-19b/PPARγ/NF-κB axis. Atherosclerosis. 2021;323:1-12. [34] YAO F, JIN Z, ZHENG Z, et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022;8(1):112. [35] GORSKI PA, JANG SP, JEONG D, et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca(2+)-ATPase in Heart Failure. Circ Res. 2019;124(9):e63-e80. [36] ZHAO T, KEE H J, BAI L, et al. Selective HDAC8 Inhibition Attenuates Isoproterenol-Induced Cardiac Hypertrophy and Fibrosis via p38 MAPK Pathway. Front Pharmacol. 2021;12:677757. [37] WANG YY, GAO B, YANG Y, et al. Histone deacetylase 3 suppresses the expression of SHP-1 via deacetylation of DNMT1 to promote heart failure. Life Sci. 2022;292: 119552. [38] RAGHUNATHAN S, GOYAL RK, PATEL BM. Selective inhibition of HDAC2 by magnesium valproate attenuates cardiac hypertrophy. Can J Physiol Pharmacol. 2017;95(3):260-267. [39] JIANG H, JIA D, ZHANG B, et al. Exercise improves cardiac function and glucose metabolism in mice with experimental myocardial infarction through inhibiting HDAC4 and upregulating GLUT1 expression. Basic Res Cardiol. 2020;115(3):28. [40] WRIGHT LH, HERR DJ, BROWN SS, et al. Angiokine Wisp-1 is increased in myocardial infarction and regulates cardiac endothelial signaling. JCI Insight. 2018;3(4):e95824. [41] TIAN S, LEI I, GAO W, et al. HDAC inhibitor valproic acid protects heart function through Foxm1 pathway after acute myocardial infarction. EBioMedicine. 2019; 39:83-94. [42] WANG Y, CHEN P, WANG L, et al. Inhibition of Histone Deacetylases Prevents Cardiac Remodeling After Myocardial Infarction by Restoring Autophagosome Processing in Cardiac Fibroblasts. Cell Physiol Biochem. 2018;49(5):1999-2011. [43] WANG K, TANG R, WANG S, et al. Isoform-Selective HDAC Inhibitor Mocetinostat (MGCD0103) Alleviates Myocardial Ischemia/Reperfusion Injury Via Mitochondrial Protection Through the HDACs/CREB/PGC-1α Signaling Pathway. J Cardiovasc Pharmacol. 2022;79(2):217-228. [44] ZHANG L, WANG H, ZHAO Y, et al. Myocyte-specific overexpressing HDAC4 promotes myocardial ischemia/reperfusion injury. Mol Med. 2018;24(1):37. [45] HUANG G, CHENG Z, HILDEBRAND A, et al. Diabetes impairs cardioprotective function of endothelial progenitor cell-derived extracellular vesicles via H3K9Ac inhibition. Theranostics. 2022;12(9):4415-4430. [46] YU Q, ZHAO J, LIU B. Bazedoxifene activates the angiotensin II-induced HUVEC hypertension model by targeting SIRT1. Exp Ther Med. 2022;23(2):120. [47] DIKALOVA A E, PANDEY A, XIAO L, et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ Res. 2020;126(4):439-452. [48] GUO J, WANG Z, WU J, et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ Res. 2019;124(10):1448-1461. [49] YOON GE, JUNG JK, LEE YH, et al. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn Schmiedebergs Arch Pharmacol. 2020;393(3):491-500. [50] DHAGIA V, KITAGAWA A, JACOB C, et al. G6PD activity contributes to the regulation of histone acetylation and gene expression in smooth muscle cells and to the pathogenesis of vascular diseases. Am J Physiol Heart Circ Physiol. 2021;320(3):H999-H1016. [51] GROOTAERT M, FINIGAN A, FIGG N L, et al. SIRT6 Protects Smooth Muscle Cells From Senescence and Reduces Atherosclerosis. Circ Res. 2021;128(4):474-491. [52] LECCE L, XU Y, V’GANGULA B, et al. Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J Clin Invest. 2021;131(15):e131178. [53] QIN J, GUO N, TONG J, et al. Function of histone methylation and acetylation modifiers in cardiac hypertrophy. J Mol Cell Cardiol. 2021;159:120-129. [54] MORIN S, CHARRON F, ROBITAILLE L, et al. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19(9):2046-2055. [55] ZHANG CL, MCKINSEY TA, CHANG S, et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479-488. [56] VEGA RB, HARRISON BC, MEADOWS E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24(19):8374-8385. [57] ZHANG J, LIANG Y, HUANG X, et al. STAT3-induced upregulation of lncRNA MEG3 regulates the growth of cardiac hypertrophy through miR-361-5p/HDAC9 axis. Sci Rep. 2019;9(1):460. [58] HU T, SCHREITER FC, BAGCHI RA, et al. HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J Biol Chem. 2019;294(21):8640-8652. [59] ZHAO X, STERNSDORF T, BOLGER TA, et al. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25(19):8456-8464. [60] SONG T, GUAN X, WANG X, et al. Dynamic modulation of gut microbiota improves post-myocardial infarct tissue repair in rats via butyric acid-mediated histone deacetylase inhibition. FASEB J. 2021;35(3):e21385. [61] LI D, YANG Y, WANG S, et al. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 2021;46:102089. [62] WU L, ZHANG Y, REN J. Epigenetic modification in alcohol use disorder and alcoholic cardiomyopathy: From pathophysiology to therapeutic opportunities. Metabolism. 2021;125:154909. [63] XU Z, SUN J, TONG Q, et al. The Role of ERK1/2 in the Development of Diabetic Cardiomyopathy. Int J Mol Sci. 2016;17(12):2001. [64] LI S J, KAO Y H, CHUNG C C, et al. HDAC I inhibitor regulates RUNX2 transactivation through canonical and non-canonical Wnt signaling in aortic valvular interstitial cells. Am J Transl Res. 2019;11(2):744-754. |
| [1] | Liu Hanfeng, Wang Jingjing, Yu Yunsheng. Artificial exosomes in treatment of myocardial infarction: current status and prospects [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 1118-1123. |
| [2] | Sun Yukang, Song Lijuan, Wen Chunli, Ding Zhibin, Tian Hao, Ma Dong, Ma Cungen, Zhai Xiaoyan. Visualization analysis of stem cell therapy for myocardial infarction based on Web of Science in recent ten years [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 1143-1148. |
| [3] | Lin Feng, Cheng Ling, Gao Yong, Zhou Jianye, Shang Qingqing. Hyaluronic acid hydrogel-encapsulated bone marrow mesenchymal stem cells promote cardiac function in myocardial infarction rats (III) [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(3): 355-359. |
| [4] | Xu Wenjie, Xie Xudong, He Ruibo, Ma Gang, Peng peng. Effect and mechanism of angiotensin (1-7) supplementation combined with exercise therapy on cardiac remodeling in rats with renal hypertension [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(26): 4137-4144. |
| [5] | Li Sijin, Feng Xiaoteng, Wang Yiru, Qin Hewei, Liu Ping. Macrophage-specific promoter SP146-C1 enhances vascular endothelial growth factor C expression in atherosclerotic mice [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(26): 4202-4208. |
| [6] | Sun Yuan, Wang Qingbo, Pi Yihua, Lu Chunmin, Xu Chuanyi, Zhang Yan. Effects of early and late aerobic exercise on right heart failure induced by monocrotaline in rats with pulmonary hypertension [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(2): 177-185. |
| [7] | Qi Ming, Wang Lei, Zhang Zhen. CXCL5 participates in carotid plaque formation by inducing vascular calcification [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(2): 186-192. |
| [8] | Sun Jinyi, Wang Qinying, Li Ying, Meng Maohua, Chen Helin, Zeng Xiao, Shu Jiayu, Li Wenjie, Luo Yuncai, Dong Qiang. The role of silent information regulator in periodontitis [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(11): 1737-1742. |
| [9] | Deng Rui, Huang Keming, Luo Jian, Chen Gong, Feng Jian, Huang Weiyi, Wei Gang. Effect of heme oxygenase-1-mediated atorvastatin on macrophage polarization and cholesterol accumulation [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(1): 62-67. |
| [10] | Zhao Quanwei, Li Hui, Liu Danan, Gong Caiwei, Chen Long. Dapagliflozin attenuates endothelial cell pyroptosis and dysfunction induced by oxidized low-density lipoprotein [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(1): 80-85. |
| [11] | Jiang Honghui, Kong Yuanyuan, Liu Jing, Wang Zhihong. Preparation and applications of decellularized extracellular matrix bioink in cardiovascular fields [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(30): 4904-4911. |
| [12] | Wang Nanfeng, Shen Shengmei, Zhang Peisheng, Teng Wei. Effects of hydrogels loaded with hepatocyte growth factor on myocardial infarction [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(30): 4802-4808. |
| [13] | Peng Fengli, Li Chaofu, Shi Bei. Application of the nanovesicle delivery system in cardiovascular diseases [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(30): 4862-4868. |
| [14] | Wei Qin, Amanguli·Ruze, Chen Bingxin, Zhao Ling, Zhao Banghao, Jiang Tao, Zhang Chun, Li Zhiqiang, Gao Xiaoming, Duan Mingjun. Relaxin protects myocardial microvascular endothelial cells from hypoxia-reoxygenation injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(28): 4519-4524. |
| [15] | Wang Shurui, Zhang Yilei, Jiao Weijie, Liu Zhihua, Zhang Kuiming, Cui Yinglin. Effects of Yiqi Tongmai Fang on serum amino acid metabolism of atherosclerotic mice [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(28): 4441-4447. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||