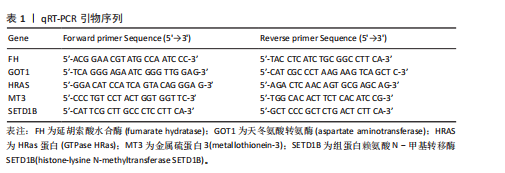
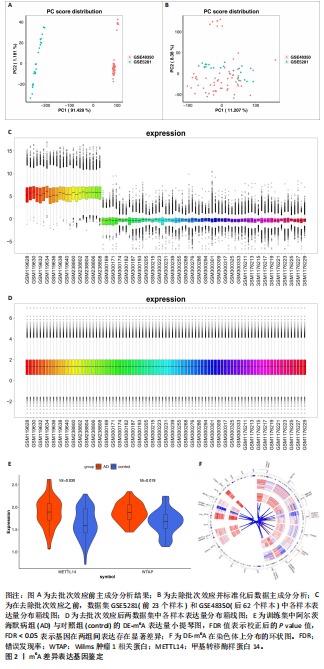
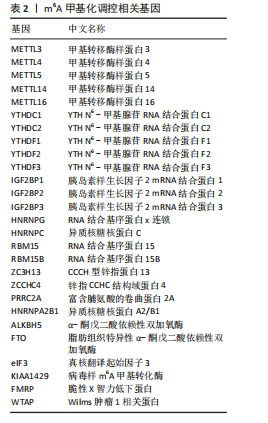
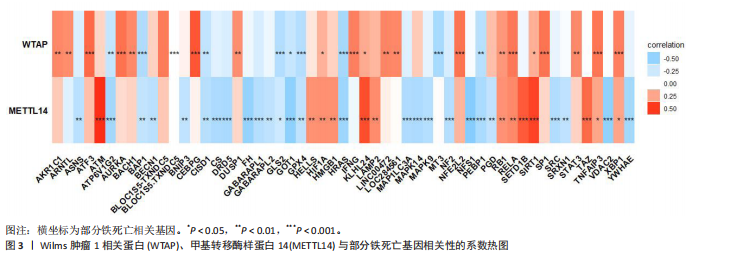
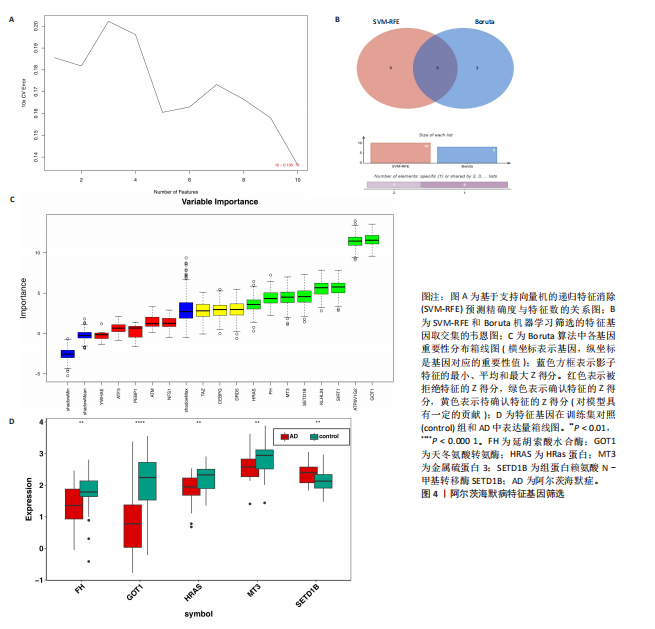
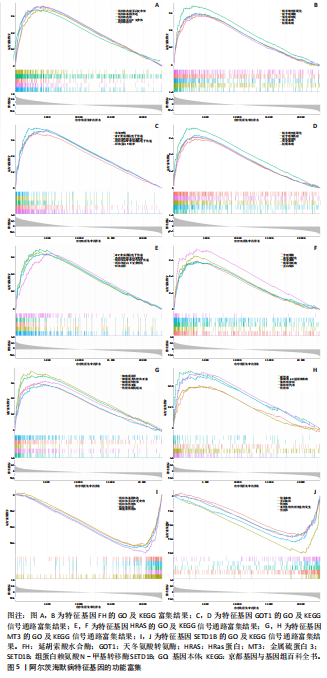
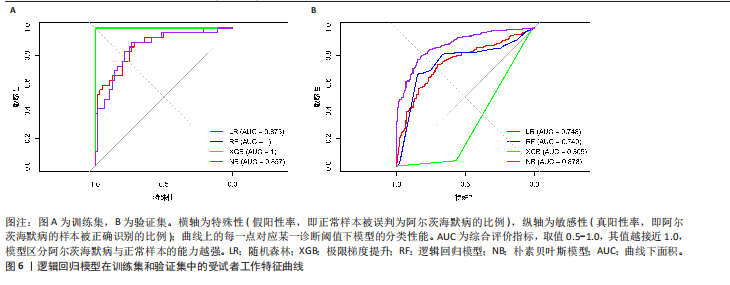
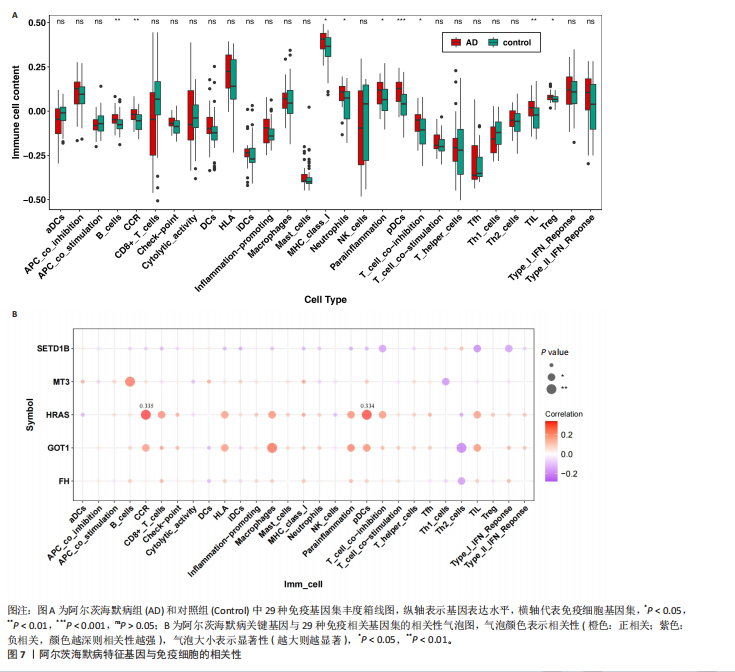
[1] LIU Y, TAN Y, ZHANG Z, et al. The interaction between ageing and Alzheimer’s disease: insights from the hallmarks of ageing. Transl Neurodegener. 2024;13(1):7.
[2] 王刚,齐金蕾,刘馨雅,等.中国阿尔茨海默病报告2024[J].诊断学理论与实践,2024, 23(3):219-256.
[3] JIA J, NING Y, CHEN M, et al. Biomarker changes during 20 years preceding Alzheimer’s disease. N Engl J Med. 2024;390(8):712-722.
[4] GRAFF-RADFORD J, YONG KXX, APOSTOLOVA LG, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021;20(3):222-234.
[5] LIU E, ZHANG Y, WANG J. Updates in Alzheimer’s disease: from basic research to diagnosis and therapies. Transl Neurodegener. 2024;13(1):45.
[6] XU Z, LV B, QIN Y, et al. Emerging roles and mechanism of m6A methylation in cardiometabolic diseases. Cells. 2022;11(7): 1101.
[7] WEI G. RNA m6A modification, signals for degradation or stabilisation? Biochem Soc Trans. 2024;52(2):707-717.
[8] HAN Y, SUN J, YAO M, et al. Biological roles of enhancer RNA m6A modification and its implications in cancer. Cell Commun Signal. 2025;23(1):254.
[9] SHAFIK AM, ZHANG F, GUO Z, et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol. 2021;22(1):17.
[10] HAN M, LIU Z, XU Y, et al. Abnormality of m6A mRNA methylation is involved in Alzheimer’s disease. Front Neurosci. 2020;14:98.
[11] ZHANG X, YANG S, HAN S, et al. Differential methylation of circRNA m6A in an APP/PS1 Alzheimer’s disease mouse model. Mol Med Rep. 2023;27(2):55.
[12] HU B, SHI Y, XIONG F, et al. Rewired m6A of promoter antisense RNAs in Alzheimer’s disease regulates neuronal genes in 3D nucleome. Nat Commun. 2025;16(1):5251.
[13] RU Q, LI Y, CHEN L, et al. Iron homeostasis and ferroptosis in human diseases: mechanisms and therapeutic prospects. Signal Transduct Target Ther. 2024;9(1):271.
[14] DIXON SJ, OLZMANN JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25: 424-442.
[15] WANG L, FANG X, LING B, et al. Research progress on ferroptosis in the pathogenesis and treatment of neurodegenerative diseases. Front Cell Neurosci. 2024;18: 1359453.
[16] ABDUKARIMOV N, KOKABI K, KUNZ J. Ferroptosis and Iron Homeostasis: Molecular Mechanisms and Neurodegenerative Disease Implications. Antioxidants (Basel). 2025; 14(5):527.
[17] LI X, CHEN J, FENG W, et al. Berberine ameliorates iron levels and ferroptosis in the brain of 3 x Tg-AD mice. Phytomedicine. 2023;118:154962.
[18] SHEN M, LI Y, WANG Y, et al. N6-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47:102151.
[19] LIU Z, LI H, PAN S. Discovery and validation of key biomarkers based on immune infiltrates in Alzheimer’s Disease. Front Genet. 2021;12: 658323.
[20] TAO X, KANG N, ZHENG Z, et al. The regulatory mechanisms of N6-methyladenosine modification in ferroptosis and its implications in disease pathogenesis. Life Sci. 2024;355: 123011.
[21] JIANG X, YAN Q, XIE L, et al. Construction and validation of a Ferroptosis-related prognostic model for gastric cancer. J Oncol. 2021;2021:6635526.
[22] HE Y, JIANG Z, CHEN C, et al. Classification of triple-negative breast cancers based on Immunogenomic profiling. J Exp Clin Cancer Res. 2018;37(1):327.
[23] 张振涛. 散发性早发型阿尔茨海默病:独特特征、关键机制与未来展望[J]. 科学通报, 2025,70(1):6-7.
[24] ZHANG R, ZHANG Y, GUO F, et al. RNA N6 -methyl‐ adenosine modifications and its roles in Alzheimer’s disease. Front Cell Neurosci. 2022;16:820378.
[25] BARBIRI I, KOUZARIDES T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020; 20(6):303-322.
[26] HAN X, GUO J, FAN Z. Interactions between m6A modifi‐ cation and miRNAs in malignant tumors. Cell Death Dis.2021;12(6):598.
[27] SENDINC E, SHI Y. RNA m6A methylation across the transcriptome. Mol Cell. 2023;83(3):428-441.
[28] MI S, SHI Y, DARI G, et al. Function of m6A and its regu lation of domesticated animals’ complex traits. J Anim Sci. 2022; 100(3):skac034.
[29] SANG A, ZHANG J, ZHANG M, et al. METTL4 mediated-N6 methyladenosine promotes acute lung injury by activating ferroptosis in alveolar epithelial cells. Free Rad Biol Med. 2024;213: 90-101.
[30] ZHU Z, HUO F, ZHANG J, et al. Crosstalk between m6A modification and alternative splicing during cancer progression. Clin Transl Med. 2023;13(10):e1460.
[31] CHEN Y, PENG C, CHEN J, et al. WTAP facilitates pro gression of hepatocellular carcinoma via m6A-HuR-depen dent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127.
[32] HUANG H, CAMATS-PERNA J, MEDEIROS R, et al. Altered expression of the m6A methyltransferase METTL3 in Alzheimer’s disease. eNeuro. 2020;7(5): ENEURO.0125-20.2020.
[33] ZHAO F, XU Y, GAO S, et al. METTL3-dependent RNA m(6)A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol Neurodegener. 2021;16(1):70.
[34] JIANG X, STOCKWELL BR, CONRAD M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021; 22(4):266-282.
[35] YANG X, ZHANG S, HE C, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19:46.
[36] FAN Z, YANG G, ZHANG W, et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J Cell Mol Med. 2021;25(21):10197-10212.
[37] WANG K, WANG G, LI G, et al. m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation. Apoptosis. 2023; 28(3-4):627-638.
[38] WANG W, CHEN J, LAI S, et al. METTL14 promotes ferroptosis in smooth muscle cells during thoracic aortic aneurysm by stabilizing the m6A modification of ACSL4. Am J Physiol Cell Physiol. 2025;328(2):C387-C399.
[39] 陈特,肖权洲,孙杨,等.METTL14介导ACSL4的m6A甲基化在髓核细胞铁死亡和细胞衰老中的作用[J].中国现代医学杂志, 2024,34(20):31-39.
[40] 宋凯.WTAP调控AR高甲基化修饰促进糖尿病心肌纤维化的分子机制[D].合肥:安徽医科大学,2024.
[41] ZHU H, LEE OW, SHAH P, et al. Identification of activators of human fumarate hydratase by quantitative high-throughput screening. SLAS Discov. 2020;25(1):43-56.
[42] 刘磊,贾少晗,于鹏.线粒体在铁死亡中的形态特征和作用[J].中国生物化学与分子生物学报,2023,39(6):769-777.
[43] PENG H, DOU H, HE S, et al. The role of GOT1 in cancer metabolism. Front Oncol. 2024; 14:1519046.
[44] ANDERSEN JV. The Glutamate/GABA-Glutamine Cycle: Insights, Updates, and Advances. J Neurochem. 2025;169(3): e70029.
[45] 赵宇翔. c-Myc通过上调GOT1和Nrf2抵抗肝癌细胞中谷氨酰胺剥夺诱发的铁死亡[D].长春:吉林大学,2020.
[46] KRMER DM, NELSON BS, LIN L, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. 2021;12: 4860.
[47] 徐箫. 综合分析铁死亡相关基因对肝细胞癌患者预后及免疫微环境的影响[D]. 青岛:青岛大学, 2023.
[48] BARTOLACCI C, ANDREANI C, EL-GAMMAL Y, et al. Lipid Metabolism Regulates Oxidative Stress and Ferroptosis in RAS-Driven Cancers: A Perspective on Cancer Progression and Therapy. Front Mol Biosci. 2021;8:706650.
[49] KOH JY, LEE SJ. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol Brain. 2020;13(1):116.
[50] GUNN AP, MCLEAN CA, CROUCH PJ, et al. Quantification of metallothionein-III in brain tissues using liquid chromatography tandem mass spectrometry. Anal Biochem. 2021;630:114326.
[51] MICHURINA A, SAKIB MS, KERIMOGLU C, et al. Postnatal expression of the lysine methyltransferase SETD1B is essential for learning and the regulation of neuron-enriched genes. EMBO J. 2022; 41(1):e106459.
[52] ZENG Z, LAN T, WEI Y, et al. CCL5/CCR5 axis in human diseases and related treatments. Genes Dis. 2022;9(1):12-27.
[53] BETTCHER BM, TANSEY MG, DOROTHÉE G, et al. Peripheral and central immune system crosstalk in Alzheimer disease - a research prospectus. Nat Rev Neurol. 2021; 17(11):689-701.
[54] PIROLLA NFF, BATISTA VS, DIAS VIGAS FP, et al. Alzheimer’s disease: related targets, synthesis of available drugs, bioactive compounds under development and promising results obtained from multi-target approaches. Curr Drug Targets. 2021; 22(5):505-538.
[55] REDDI SREE R, KALYAN M, ANAND N, et al. Newer Therapeutic Approaches in Treating Alzheimer’s Disease: A Comprehensive Review. ACS Omega. 2025;10(6):5148-5171.
|