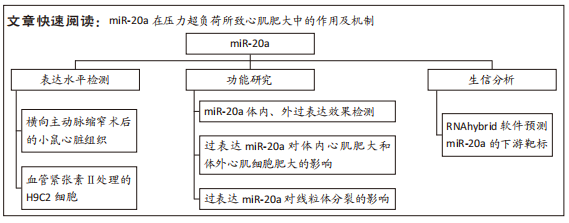
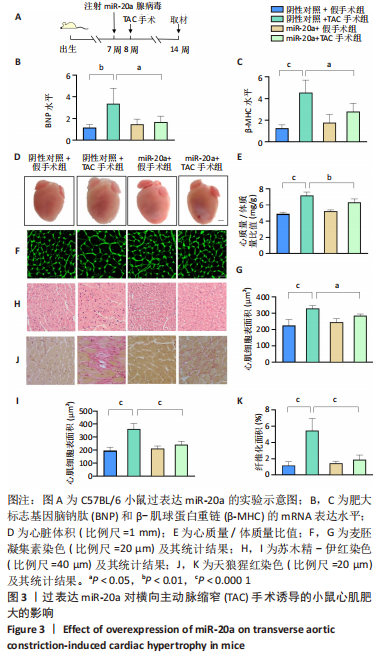
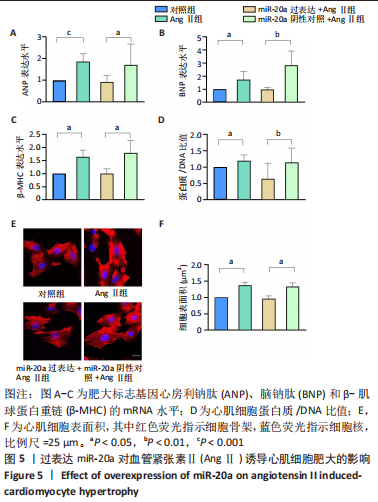
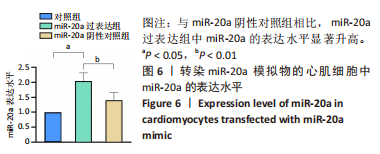
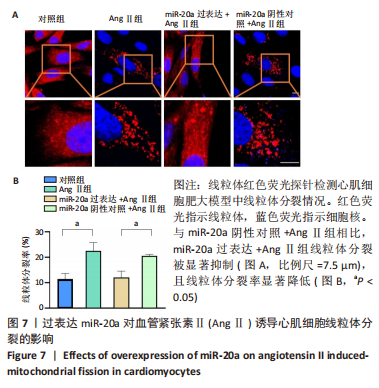
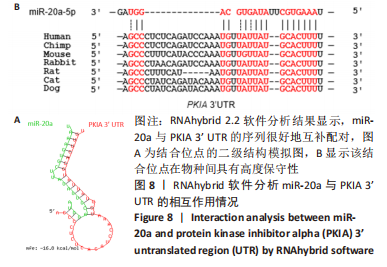
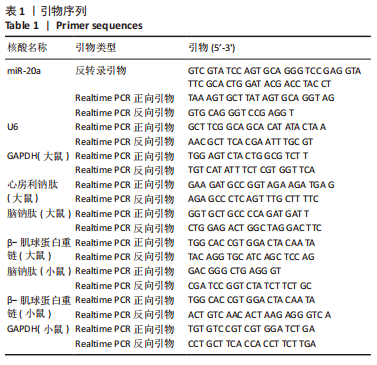
[1] NAKAMURA M, SADOSHIMA J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387-407.
[2] KUWAHARA K, NISHIKIMI T, NAKAO K. Transcriptional regulation of the fetal cardiac gene program. J Pharmacol Sci. 2012;119(3):198-203.
[3] WANG K, LONG B, LIU F, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37(33): 2602-2611.
[4] TONG XH, LIU QH, XU SH, et al. Changes in RNA, DNA, protein contents and growth of turbot Scophthalmus maximus larvae and juveniles. J Fish Biol. 2010; 77(3):512-525.
[5] YANG J, CHEN Y, LI X, et al. New insights into the roles of glucocorticoid signaling dysregulation in pathological cardiac hypertrophy. Heart Fail Rev. 2022;27(4): 1431-1441.
[6] LI X, TAN W, ZHENG S, et al. Differential mRNA Expression and Circular RNA-Based Competitive Endogenous RNA Networks in the Three Stages of Heart Failure in Transverse Aortic Constriction Mice. Front Physiol. 2022;13:777284.
[7] JIN JY, WEI XX, ZHI XL, et al. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. 2021;42(5):655-664.
[8] WAI T, GARCÍA-PRIETO J, BAKER MJ, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science. 2015; 350(6265):aad0116.
[9] FORTE M, SCHIRONE L, AMERI P, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. 2021;178(10):2060-2076.
[10] AUNG LHH, JUMBO JCC, WANG Y, et al. Therapeutic potential and recent advances on targeting mitochondrial dynamics in cardiac hypertrophy: A concise review. Mol Ther Nucleic Acids. 2021;25:416-443.
[11] LIN J, DUAN J, WANG Q, et al. Mitochondrial Dynamics and Mitophagy in Cardiometabolic Disease. Front Cardiovasc Med. 2022;9:917135.
[12] RUPAIMOOLE R, SLACK FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203-222.
[13] LI J, SHA Z, ZHU X, et al. Targeting miR-30d reverses pathological cardiac hypertrophy. EBioMedicine. 2022;81:104108.
[14] UCAR A, GUPTA SK, FIEDLER J, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078.
[15] GABISONIA K, PROSDOCIMO G, AQUARO GD, et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature. 2019; 569(7756):418-422.
[16] HE D, HU J, LU Y, et al. The Effect of miR-505-5p on Inhibition of Serum Uromodulin Ameliorates Myocardial Inflammation and Apoptosis Induced by Ischemia-Reperfusion. Oxid Med Cell Longev. 2022;2022:3521971.
[17] DENG Z, YAO J, XIAO N, et al. DNA methyltransferase 1 (DNMT1) suppresses mitophagy and aggravates heart failure via the microRNA-152-3p/ETS1/RhoH axis. Lab Invest. 2022;102(8):782-793.
[18] YANG Y, WANG Z, XU Y, et al. Knockdown of lncRNA H19 alleviates ox-LDL-induced HCAECs inflammation and injury by mediating miR-20a-5p/HDAC4 axis. Inflamm Res. 2022;71(9):1109-1121.
[19] WANG D, WANG Y, MA J, et al. MicroRNA-20a participates in the aerobic exercise-based prevention of coronary artery disease by targeting PTEN. Biomed Pharmacother. 2017;95:756-763.
[20] SHEU JJ, CHAI HT, SUNG PH, et al. Double overexpression of miR-19a and miR-20a in induced pluripotent stem cell-derived mesenchymal stem cells effectively preserves the left ventricular function in dilated cardiomyopathic rat. Stem Cell Res Ther. 2021;12(1):371.
[21] LIU X, GUO B, ZHANG W, et al. MiR-20a-5p overexpression prevented diabetic cardiomyopathy via inhibition of cardiomyocyte apoptosis, hypertrophy, fibrosis and JNK/NF-κB signalling pathway. J Biochem. 2021;170(3):349-362.
[22] AN D, ZENG Q, ZHANG P, et al. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox Biol. 2021;46:102088.
[23] JU S, PARK S, LIM L, et al. Low density lipoprotein receptor-related protein 1 regulates cardiac hypertrophy induced by pressure overload. Int J Cardiol. 2020; 299:235-242.
[24] WANG T, ZHAI M, XU S, et al. NFATc3-dependent expression of miR-153-3p promotes mitochondrial fragmentation in cardiac hypertrophy by impairing mitofusin-1 expression. Theranostics. 2020;10(2):553-566.
[25] WANG K, LONG B, JIAO JQ, et al. miR-484 regulates mitochondrial network through targeting Fis1. Nat Commun. 2012;3:781.
[26] WANG K, LONG B, ZHOU LY, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. 2014;5:3596.
[27] MCGEARY SE, LIN KS, SHI CY, et al. The biochemical basis of microRNA targeting efficacy. Science. 2019;366(6472):eaav1741.
[28] REHMSMEIER M, STEFFEN P, HOCHSMANN M, et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507-1517.
[29] ZHANG F, ZHANG L, QI Y, et al. Mitochondrial cAMP signaling. Cell Mol Life Sci. 2016;73(24):4577-4590.
[30] SOTOMAYOR-FLORES C, RIVERA-MEJÍAS P, VÁSQUEZ-TRINCADO C, et al. Angiotensin-(1-9) prevents cardiomyocyte hypertrophy by controlling mitochondrial dynamics via miR-129-3p/PKIA pathway. Cell Death Differ. 2020; 27(9):2586-2604.
[31] LUO Y, JIANG N, MAY HI, et al. Cooperative Binding of ETS2 and NFAT Links Erk1/2 and Calcineurin Signaling in the Pathogenesis of Cardiac Hypertrophy. Circulation. 2021;144(1):34-51.
[32] LIN QY, ZHANG YL, BAI J, et al. VEGF-C/VEGFR-3 axis protects against pressure-overload induced cardiac dysfunction through regulation of lymphangiogenesis. Clin Transl Med. 2021;11(3):e374.
[33] BERNARDO BC, WEEKS KL, PRETORIUS L, et al. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128(1):191-227.
[34] CHENG Y, SHEN A, WU X, et al. Qingda granule attenuates angiotensin II-induced cardiac hypertrophy and apoptosis and modulates the PI3K/AKT pathway. Biomed Pharmacother. 2021;133:111022.
[35] TAKEZAKO T, UNAL H, KARNIK SS, et al. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol Res. 2017;123:40-50.
[36] ROSENBAUGH EG, SAVALIA KK, MANICKAM DS, et al. Antioxidant-based therapies for angiotensin II-associated cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol. 2013;304(11):R917-928.
[37] LIN HB, NAITO K, OH Y, et al. Innate Immune Nod1/RIP2 Signaling Is Essential for Cardiac Hypertrophy but Requires Mitochondrial Antiviral Signaling Protein for Signal Transductions and Energy Balance. Circulation. 2020;142(23):2240-2258.
[38] PIQUEREAU J, CAFFIN F, NOVOTOVA M, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res. 2012;94(3):408-417.
[39] KONG AS, LAI KS, LIM SE, et al. miRNA in Ischemic Heart Disease and Its Potential as Biomarkers: A Comprehensive Review. Int J Mol Sci. 2022;23(16):9001.
[40] GARGIULO P, MARZANO F, SALVATORE M, et al. MicroRNAs: diagnostic, prognostic and therapeutic role in heart failure-a review. ESC Heart Fail. 2022 Nov 8. doi: 10.1002/ehf2.14153. Online ahead of print.
[41] PARVAN R, HOSSEINPOUR M, MORADI Y, et al. Diagnostic performance of microRNAs in the detection of heart failure with reduced or preserved ejection fraction: a systematic review and meta-analysis. Eur J Heart Fail. 2022;24(12):2212-2225.
[42] LUO F, LIU W, BU H. MicroRNAs in hypertrophic cardiomyopathy: pathogenesis, diagnosis, treatment potential and roles as clinical biomarkers. Heart Fail Rev. 2022;27(6):2211-2221.
[43] ZHANG Y, WANG Z, GEMEINHART RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962-974.
[44] TÄUBEL J, HAUKE W, RUMP S, et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur Heart J. 2021;42(2):178-188.
[45] HU L, DING M, TANG D, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. 2019;9(13):3687-3706.
[46] ZHAO Y, PONNUSAMY M, LIU C, et al. MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(11):2871-2881.
[47] ZHOU L, LI R, LIU C, et al. Foxo3a inhibits mitochondrial fission and protects against doxorubicin-induced cardiotoxicity by suppressing MIEF2. Free Radic Biol Med. 2017;104:360-370.
[48] QI J, WANG F, YANG P, et al. Mitochondrial Fission Is Required for Angiotensin II-Induced Cardiomyocyte Apoptosis Mediated by a Sirt1-p53 Signaling Pathway. Front Pharmacol. 2018;9:176.
|