中国组织工程研究 ›› 2014, Vol. 18 ›› Issue (29): 4675-4682.doi: 10.3969/j.issn.2095-4344.2014.29.015
• 组织构建与生物活性因子 tissue construction and bioactive factors • 上一篇 下一篇
人胶质细胞源性神经营养因子和血管内皮生长因子165双基因真核表达载体的构建与鉴定
栗炳南,李卫东,林俊堂,丰慧根
- 新乡医学院生命科学技术学院,河南省新乡市 453003
-
修回日期:2014-05-01出版日期:2014-07-09发布日期:2014-07-09 -
通讯作者:栗炳南,新乡医学院生命科学技术学院,河南省新乡市 453003 -
作者简介:栗炳南,男,1983年生,河南省新乡市人,汉族,2011年中南大学湘雅医学院毕业,博士,讲师,目前主要从事神经系统疾病的发病机制与治疗方面的研究。 并列第一作者:李卫东,新乡医学院生命科学技术学院,河南省新乡市 453003 -
基金资助:新乡医学院重点领域招标课题(ZD2011-16);河南省教育厅科学技术研究重点项目(13A180850)
Construction and identification of pIRES2-GDNF-VEGF165 bicistronic eukaryotic expression vector
Li Bing-nan, Li Wei-dong, Lin Jun-tang, Feng Hui-gen
- Department of Life Sciences and Technology, Xinxiang Medical University, Xinxiang 453003, Henan Province, China
-
Revised:2014-05-01Online:2014-07-09Published:2014-07-09 -
Contact:Li Bing-nan, Department of Life Sciences and Technology, Xinxiang Medical University, Xinxiang 453003, Henan Province, China -
About author:Li Bing-nan, Ph.D., Lecturer, Department of Life Sciences and Technology, Xinxiang Medical University, Xinxiang 453003, Henan Province, China Li Wei-dong, Department of Life Sciences and Technology, Xinxiang Medical University, Xinxiang 453003, Henan Province, China Li Bing-nan and Li Wei-dong contributed equally to this work. -
Supported by:the Tender Subject of Key Research Areas of Xinxiang Medical University in 2011, No. ZD2011-16; Key Projects in Scientific Research of Henan Provincial Education Department, No. 13A180850.
摘要:
背景:人胶质细胞源性神经营养因子(glial cell line - derived neurotrophic factor,GDNF)和血管内皮生长因子165 (vascular endothelial growth factor 165,VEGF165)在细胞分化过程中有重要作用。
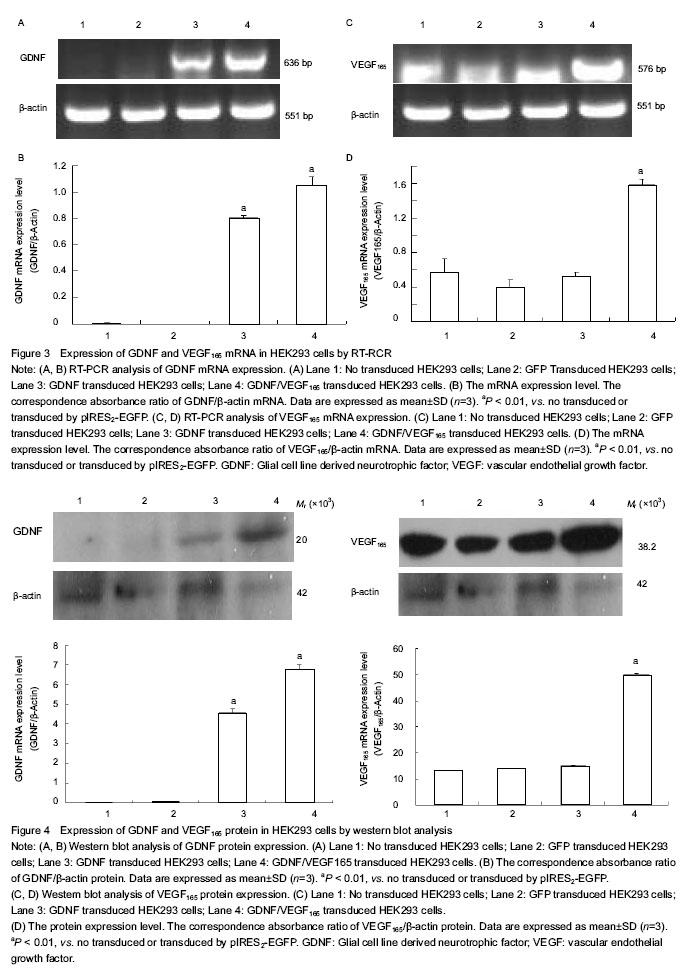
结果与结论:DNA测序显示,提取的人胶质细胞源性神经营养因子和血管内皮生长因子165均与基因库报道序列一致,相对分子质量分别为636 bp和576 bp。构建的pIRES2-GDNF-VEGF165双基因共表达载体经Bgl II/Bam HI切出GDNF条带,经Bam HI/Not I双酶切后切出 IRES-VEGF165片段,经Bgl II/Not I双酶切后切出GDNF-IRES-VEGF165片段。RT-PCR与Western-blot方法检测显示,此载体转染后,HEK293细胞均能表达人胶质细胞源性神经营养因子和血管内皮生长因子165 mRNA和蛋白。说明实验成功构建了能表达人胶质细胞源性神经营养因子和血管内皮生长因子165的双基因真核表达载体。
中图分类号:
引用本文
栗炳南,李卫东,林俊堂,丰慧根. 人胶质细胞源性神经营养因子和血管内皮生长因子165双基因真核表达载体的构建与鉴定[J]. 中国组织工程研究, 2014, 18(29): 4675-4682.
Li Bing-nan, Li Wei-dong, Lin Jun-tang, Feng Hui-gen. Construction and identification of pIRES2-GDNF-VEGF165 bicistronic eukaryotic expression vector[J]. Chinese Journal of Tissue Engineering Research, 2014, 18(29): 4675-4682.
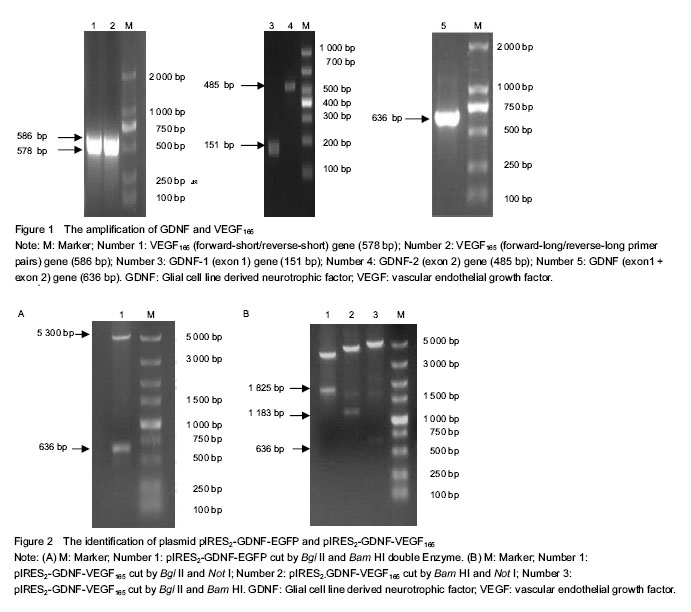
| [1] Zurita M, Aguayo C, Bonilla C, et al. The pig model of chronic paraplegia: a challenge for experimental studies in spinal cord injury. Prog Neurobiol. 2012;97(3):288-303. [2] Shah AM, Mann DL. In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet. 2011;378(9792):704-712. [3] Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873-887. [4] Fournier NM1, Duman RS. Role of vascular endothelial growth factor in adult hippocampal neurogenesis: implications for thepathophysiology and treatment of depression. Behav Brain Res. 2012;227(2):440-449. [5] Gu X, Ding F, Yang Y, et al. Construction of tissue engineered nerve grafts and their application in peripheral nerve regeneration. Prog Neurobiol. 2011;93(2):204-230. [6] Warner-Schmidt JL, Duman RS. VEGF as a potential target for therapeutic intervention in depression. Curr Opin Pharmacol. 2008;8(1):14-19. [7] Kubota H, Wu X, Goodyear SM, et al. Glial cell line-derived neurotrophic factor and endothelial cells promote self-renewal of rabbit germ cells withspermatogonial stem cell properties. FASEB J. 2011;25(8):2604-2614. [8] Savitt J, Singh D, Zhang C, et al. The in vivo response of stem and other undifferentiated spermatogonia to the reversible inhibition of glial cell line-derived neurotrophic factor signaling in the adult. Stem Cells. 2012;30(4): 732-740. [9] May F, Buchner A, Schlenker B, et al. Schwann cell-mediated delivery of glial cell line-derived neurotrophic factor restores erectile function aftercavernous nerve injury. Int J Urol. 2013;20(3):344-348. [10] Rodrigues DM, Li AY, Nair DG, et al. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol Motil. 2011; 23(2):e44-56. [11] Stahl K, Mylonakou MN, Skare O, et al. Cytoprotective effects of growth factors: BDNF more potent than GDNF in an organotypic culture model of Parkinson's disease. Brain Res. 2011;1378(1):105-118. [12] Madduri S1, Feldman K, Tervoort T, et al. Collagen nerve conduits releasing the neurotrophic factors GDNF and NGF. J Control Release. 2010;143(2):168-174. [13] Lin W, Li M, Li Y, et al. Bone marrow stromal cells promote neurite outgrowth of spinal motor neurons by means of neurotrophic factorsin vitro. Neurol Sci. 2014;35(3): 449-457. [14] Deng LX, Deng P, Ruan Y, et al. A novel growth-promoting pathway formed by GDNF-overexpressing Schwann cells promotes propriospinalaxonal regeneration, synapse formation, and partial recovery of function after spinal cord injury. J Neurosci. 2013;33(13):5655-5667. [15] Gyorkos AM, McCullough MJ, Spitsbergen JM. Glial cell line derived neurotrophic factor (GDNF) expression and NMJ plasticity in skeletal muscle followingendurance exercise. Neuroscience. 2014;257(1):111-118. [16] Newland B, Abu-Rub M, Naughton M, et al. GDNF gene delivery via a 2-(dimethylamino) ethyl methacrylate based cyclized knot polymer for neuronal cellapplications. ACS Chem Neurosci. 2013;4(4):540-546. [17] Nikkhah G. Restorative strategies for the dopaminergic nigrostriatal projection pathway. Acta Neurochir Suppl. 2013;117:79-85. [18] Yu ZQ, Zhang BL, Ren QX, Changes in transcriptional factor binding capacity resulting from promoter region methylation induce aberrantlyhigh GDNF expression in human glioma. Mol Neurobiol. 2013;48(3):571-580. [19] Xie BH, Xie YF. Twin PCRs: a simple and efficient method for directional cloning of PCR products. World J Microbiol Biotechnol. 2011;27(9):2223-2225. [20] Chou AK, Yang MC, Tsai HP, et al. Adenoviral-mediated glial cell line-derived neurotrophic factor gene transfer has a protective effect on sciatic nerve following constriction-induced spinal cord injury. PLoS One. 2014; 9(3): e92264. [21] Kubota H, Wu X, Goodyear SM, et al. Glial cell line-derived neurotrophic factor and endothelial cells promote self-renewal of rabbit germ cells with spermatogonial stem cell properties. FASEB J. 2011;25(8):2604-2614. [22] Savitt J, Singh D, Zhang C, et al. The in vivo response of stem and other undifferentiated spermatogonia to the reversible inhibition of glial cell line-derived neurotrophic factor signaling in the adult. Stem Cells. 2012;30(4): 732-740. [23] May F, Buchner A, Schlenker B, et al. Schwann cell-mediated delivery of glial cell line-derived neurotrophic factor restores erectile function after cavernous nerve injury. Int J Urol. 2013;20(3):344-348. [24] Fletcher AM, Kowalczyk TH, Padegimas L, et al. Transgene expression in the striatum following intracerebral injections of DNA nanoparticles encoding for human glial cell line-derived neurotrophic factor. Neuroscience. 2011; 194: 220-226. [25] Rodrigues DM, Li AY, Nair DG, et al. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol Motil. 2011; 23(2):e44-56. [26] Xu P, Rosen KM, Hedstrom K, et al. Nerve injury induces glial cell line-derived neurotrophic factor (GDNF) expression in Schwann cells throughpurinergic signaling and the PKC-PKD pathway. Glia. 2013;61(7):1029-1040. [27] Nowacka MM, Obuchowicz E. Vascular endothelial growth factor (VEGF) and its role in the central nervous system: a new element in the neurotrophic hypothesis of antidepressant drug action. Neuropeptides. 2012; 46(1): 1-10. [28] Luo H, Zhang Y, Zhang Z, et al. The protection of MSCs from apoptosis in nerve regeneration by TGFβ1 through reducing inflammation andpromoting VEGF-dependent angiogenesis. Biomaterials. 2012;33(17):4277-4287. [29] Mackenzie F, Ruhrberg C. Diverse roles for VEGF-A in the nervous system. Development. 2012;139(8):1371-1380. [30] Li Z, Burns AR, Han L, et al. IL-17 and VEGF are necessary for efficient corneal nerve regeneration. Am J Pathol. 2011; 178(3):1106-1116. |
| [1] | 冉江华,刘 静,张熙冰,张升宁,吴淑媛,李来邦,李 望,李 立. 恒河猴肝移植模型急性排斥反应时T细胞亚群的变化[J]. 中国组织工程研究, 2014, 18(49): 7948-7954. |
| [2] | 郑鳕洋,韩 澍,周梅生,傅尚希,王立明. 羟苯磺酸钙治疗慢性移植肾功能不全的临床观察[J]. 中国组织工程研究, 2014, 18(49): 7979-7984. |
| [3] | 麻海亮,于新波,刘梦东,贾 婧,符大勇. 恒牙牙体硬组织钙磷成分的检测:分光光度法与EDTA滴定法[J]. 中国组织工程研究, 2014, 18(46): 7432-7436. |
| [4] | 刘长剑,王 李,罗宗键. 低能体外冲击波和低剂量间歇甲状旁腺素干预成骨细胞的增殖和分化[J]. 中国组织工程研究, 2014, 18(11): 1672-1679. |
| [5] | 麦志辉,张静兰,卢红飞,陈 奇,梁焕友,艾 虹. 牙周膜牵引成骨快速远中移动尖牙的可行性[J]. 中国组织工程研究, 2013, 17(41): 7255-7264. |
Design
.jpg)
.jpg)
在基因治疗中,联合应用多个具有协同作用的治疗基因通常可产生较单基因更为理想的效果。但是与国内外同类研究水平的比较以往进行多基因联合治疗的方法存在不同缺陷,利用携带不同基因的独立载体系统同时转染靶细胞的方法,虽然可自由调节各表达载体的比例,但各基因的总表达效率低下;而用传统方法构建的多启动子表达载体,因为每个治疗基因都需要一个包括启动子、终止子等表达元件在内的完整基因表达盒,使得载体过于庞大,操作困难;而2个基因融合表达的策略可能因蛋白结构相互影响而导致功能丧失。利用IRES构建多基因共表达载体,则可大大提高转移及表达效率。实验将人胶质细胞源性神经营养因子和血管内皮生长因子165双基因成功插入到了双顺反子真核表达载体pIRES2中,采用一种简便和高效的方法成功构建了pIRES2-人胶质细胞源性神经营养因子-血管内皮生长因子165双基因共表达载体。
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||