Chinese Journal of Tissue Engineering Research ›› 2024, Vol. 28 ›› Issue (21): 3400-3406.doi: 10.12307/2024.082
Previous Articles Next Articles
Regulation of bone tissue cells by tumor necrosis factor-alpha
Wang Xin, Wubulikasimu·Mijiti, Huang Jinyong, Xie Zengru
- Department of Orthopedics and Trauma, First Affiliated Hospital of Xinjiang Medical University, Urumqi 830011, Xinjiang Uygur Autonomous Region, China
-
Received:2023-05-27Accepted:2023-07-10Online:2024-07-28Published:2023-09-28 -
Contact:Xie Zengru, Chief physician, Professor, Department of Orthopedics and Trauma, First Affiliated Hospital of Xinjiang Medical University, Urumqi 830011, Xinjiang Uygur Autonomous Region, China -
About author:Wang Xin, Doctoral candidate, Physician, Department of Orthopedics and Trauma, First Affiliated Hospital of Xinjiang Medical University, Urumqi 830011, Xinjiang Uygur Autonomous Region, China Wubulikasimu·Mijiti, Department of Orthopedics and Trauma, First Affiliated Hospital of Xinjiang Medical University, Urumqi 830011, Xinjiang Uygur Autonomous Region, China -
Supported by:National Natural Science Foundation of China, No. 82260409 (to XZR); Key Project of Science and Technology Department of Xinjiang Uygur Autonomous Region, No. 2021D01D19 (to XZR); Postgraduate Practice Innovation Project of the Autonomous Region, No. XJ2023G165 (to WX)
CLC Number:
Cite this article
Wang Xin, Wubulikasimu·Mijiti, Huang Jinyong, Xie Zengru. Regulation of bone tissue cells by tumor necrosis factor-alpha[J]. Chinese Journal of Tissue Engineering Research, 2024, 28(21): 3400-3406.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks

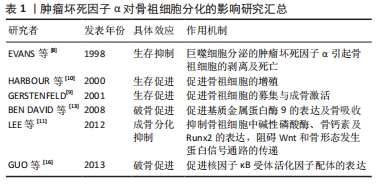
2.1 肿瘤坏死因子α的产生、受体及作用途径 肿瘤坏死因子α基因位于人类的第6号染色体(小家鼠的第17号染色体,褐家鼠的20号染色体)上,该基因由4个外显子和3个内含子组成。肿瘤坏死因子α mRNA翻译后生成相对分子质量27 000的蛋白质,该蛋白与细胞膜紧密结合,称为膜结合肿瘤坏死因子α。膜结合肿瘤坏死因子α通过名为肿瘤坏死因子α 转换酶(TACE)的水解蛋白酶切割后形成相对分子质量17 000的可溶性肿瘤坏死因子α。一般认为膜结合肿瘤坏死因子α以自分泌/旁分泌方式调节生物反应,而可溶性肿瘤坏死因子α通过内分泌方式引起效应[3]。 肿瘤坏死因子α可在全身多种细胞中表达和分泌,具有吞噬功能的单核/巨噬细胞系统是肿瘤坏死因子α的主要来源。部分免疫细胞,如CD4+淋巴细胞、B细胞、肥大细胞及自然杀伤细胞也分泌肿瘤坏死因子α。而在骨组织中,已知成骨细胞及其前体间充质干细胞、破骨细胞及其前体巨噬细胞细胞均可表达肿瘤坏死因子α。 肿瘤坏死因子α可与两种细胞表面肿瘤坏死因子受体结合,即肿瘤坏死因子1型受体和肿瘤坏死因子2型受体。通常认为肿瘤坏死因子1型受体与肿瘤坏死因子α的结合是不可逆的,而肿瘤坏死因子2型受体与肿瘤坏死因子α可以快速的结合及解离。肿瘤坏死因子1型受体可被可溶性和膜结合形式有效激活,而肿瘤坏死因子2型受体主要以膜结合形式激活,与肿瘤坏死因子α亲和力较低。肿瘤坏死因子1型受体的胞质内部分含有死亡结构域,死亡结构域介导细胞毒效应,因此肿瘤坏死因子1型受体主要介导细胞凋亡,参与免疫炎症反应。而肿瘤坏死因子2型受体不含死亡结构域,主要影响细胞的增殖、生存及活化[4-5]。肿瘤坏死因子1型受体与肿瘤坏死因子2型受体的结构域和效应之间存在复杂的相互作用。肿瘤坏死因子1型受体广泛分布于几乎所有种类细胞的胞膜表面,而肿瘤坏死因子2型受体主要表达于在免疫细胞、内皮细胞、神经细胞和各类干细胞表面。目前认为骨细胞、巨噬细胞及破骨细胞同时表达肿瘤坏死因子1型和2型受体,但有关成骨细胞及间充质干细胞是否表达肿瘤坏死因子2型受体仍存争议[4]。相关研究通过对相关受体靶基因进行沉默,发现肿瘤坏死因子2型受体对成骨细胞分化起显著影响[6],故文章倾向于认为成骨细胞及间充质干细胞同时表达肿瘤坏死因子1型和2型受体。 2.2 肿瘤坏死因子α对骨祖细胞的影响 骨祖细胞也称为前骨母细胞,来源于具有多种分化潜力的间充质干细胞,起源于神经外胚层及中胚层。位于骨皮质内及骨膜下,具有分化为成骨细胞及软骨细胞的能力[7]。近些年来,由于处于成骨分化体系中的细胞缺乏清晰的界限,骨祖细胞在体外模型中的界定也较为模糊,故针对肿瘤坏死因子α对骨祖细胞影响的研究也较少。但就目前已有的文献报道来看,骨祖细胞在肿瘤坏死因子α的影响下偏向于生存抑制、成骨分化抑制及生存促进。 2.2.1 肿瘤坏死因子α调控骨祖细胞的活性与成骨分化 相关研究指出,如在松动假体组织等特定组织环境下,植入物碎片诱导巨噬细胞表达的肿瘤坏死因子α可直接引起骨祖细胞的剥离及死亡[8]。但也有研究指出肿瘤坏死因子α参与了膜内成骨过程中骨祖细胞的募集[9],或促进骨祖细胞的增殖[10]。此外,虽然既往研究已经提出肿瘤坏死因子α、白细胞介素1β和白细胞介素6等促炎细胞因子均可抑制成骨细胞的分化,但只有肿瘤坏死因子α能够抑制骨祖细胞中的碱性磷酸酶活性,并同时抑制了骨钙素和Runx2的表达,进而减少骨组织基质矿化和胶原合成。上述研究认为肿瘤坏死因子α在炎性细胞因子对骨祖细胞成骨分化抑制中的重要地位[11]。此外,肿瘤坏死因子α还可通过直接调节Wnt和骨形态发生蛋白信号通路,或诱导SOST的自分泌,通过阻碍Wnt和骨形态发生蛋白信号通路的传递来阻碍骨祖细胞的成骨分化过程[12]。 2.2.2 肿瘤坏死因子α促进骨祖细胞参与骨吸收及破骨分化 有研究提出骨组织细胞中除了破骨细胞具有骨吸收能力外,暴露于肿瘤坏死因子α的骨祖细胞也可通过分泌基质金属蛋白酶9,降解骨基质中的胶原蛋白和基质蛋白,主动参与骨吸收过程[13-14]。此效应可能与骨祖细胞中核转录因子κB通路的激活有关[15]。此外,肿瘤坏死因子α 通过刺激骨祖细胞表达核因子κB受体活化因子配体促进破骨细胞分化[16]。 文章总结了肿瘤坏死因子α对骨祖细胞分化的影响研究进展,见表1。"
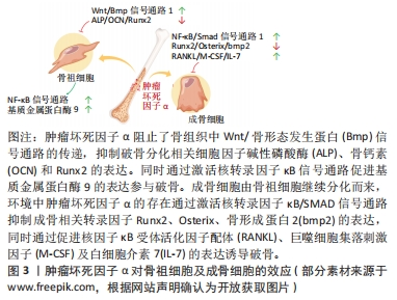
2.3 肿瘤坏死因子α对破骨细胞的影响 肿瘤坏死因子α可通过多种方式作用于破骨及其前体细胞,发挥促破骨效应,主要包括:①增加破骨前体细胞表面促破骨受体的表达;②增强对促破骨细胞因子的敏感性;③激活破骨相关信号通路;④促进成骨系细胞表达促破骨细胞因子;⑤肿瘤坏死因子α与特定种类的细胞因子配合直接促进破骨细胞的生成。破骨细胞作为唯一具有明确骨吸收功能的骨组织细胞,肿瘤坏死因子α的促进效应将使成骨-破骨平衡向破骨方向倾斜。 2.3.1 破骨细胞的分化途径与功能 破骨细胞是骨组织中唯一的一种骨吸收细胞,参与骨组织的改造重塑。早期学界认为,破骨细胞来源于骨髓内的造血干细胞,经造血干细胞-多能祖细胞-髓共同祖细胞-破骨祖细胞的顺序逐步分化,最终破骨祖细胞在细胞因子的作用下融合形成成熟的破骨细胞。最近研究证明,炎症环境下的单核/巨噬细胞也是破骨细胞的重要来源[17]。 2.3.2 肿瘤坏死因子α通过激活相关信号通路促进破骨细胞分化 核因子κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)是激活破骨细胞分化的主要细胞因子。RANKL与肿瘤坏死因子α同属肿瘤坏死因子超家族,RANKL受体核因子κB受体活化因子(receptor activator of nuclear factor-κB,RANK)与TNFR同属肿瘤坏死因子受体超家族。包括RANK在内的大多数肿瘤坏死因子受体家族成员与一个称为肿瘤坏死因子受体相关因子的衔接蛋白家族相互作用。当肿瘤坏死因子α与肿瘤坏死因子1型受体结合后,与TRADD、RIP1和肿瘤坏死因子受体相关因子2组合形成蛋白复合物,该复合物激活蛋白激酶复合物IKKs,IKKs随后磷酸化、泛素化并降解IκB蛋白,将核转录因子κB从核转录因子κB/IκB复合物中分离并转移到细胞核内,结合到目标基因启动子区域,启动目标基因的转录[18]。而核转录因子κB信号通路被认为是破骨细胞形成过程中的关键信号通路之一[19]。在已知的肿瘤坏死因子受体相关因子家族成员中,肿瘤坏死因子受体相关因子6在破骨细胞分化中起着关键作用。虽然肿瘤坏死因子1型/2型受体均不衔接肿瘤坏死因子受体相关因子6,但肿瘤坏死因子α诱导肿瘤坏死因子受体相关因子2 信号的表达,肿瘤坏死因子受体相关因子2通过增强 RANK?肿瘤坏死因子受体相关因子6信号通路促进破骨细胞形成[19]。 2.3.3 肿瘤坏死因子α增强RANK/RANKL的表达和效应促进破骨细胞生成 肿瘤坏死因子α可通过环氧合酶/前列腺素 E 途径及促进活化T细胞核因子1/环磷腺苷效应元件结合蛋白与RANKL启动子结合的方式诱导成骨类细胞表达RANKL[20]。此外,当环境内RANKL存在时,肿瘤坏死因子α以剂量依赖性方式增强破骨细胞分化。即使RANKL浓度不足其单独诱导所需浓度的1%,也可以在肿瘤坏死因子α的协助下稳定的进行破骨分化。其机制为肿瘤坏死因子α和RANKL协同激活了应激活化蛋白激酶/c-Jun氨基末端激酶和核转录因子κB信号通路[21-24]。当体内炎症反应严重时,较高浓度的肿瘤坏死因子α通过直接诱导巨噬细胞RANK的表达促进破骨,而在长期慢性的炎症环境中,较低浓度的肿瘤坏死因子α主要通过促使骨髓间充质干细胞等除巨噬细胞以外的细胞分泌RANKL和巨噬细胞集落刺激因子来促进破骨。如果炎症反应逐渐加重,肿瘤坏死因子α浓度增高,则同样促进破骨前体细胞中RANK和集落刺激因子1受体的表达[25]。此时RANKL的表达的增加不再重要,因为正常生理水平的RANKL已经能够导致破骨增加[26-28]。此外,有研究提及肿瘤坏死因子α诱导骨髓间充质干细胞合成RANKL,RANKL也通过激活肿瘤坏死因子α启动子中的 核转录因子κB 位点促进破骨前体细胞中肿瘤坏死因子α mRNA 的转录[29],提示骨组织中肿瘤坏死因子α与RANKL的表达可能存在相互促进效应,见图3。"
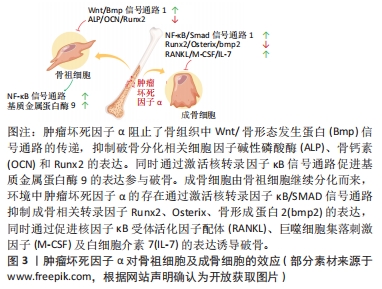
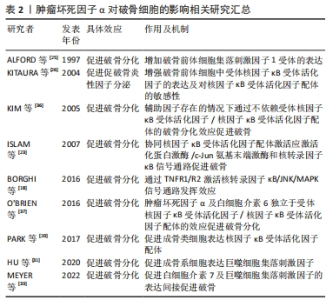
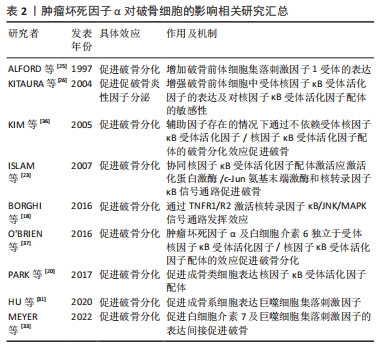
2.3.4 肿瘤坏死因子α通过刺激巨噬细胞集落刺激因子的产生促进破骨细胞生成 肿瘤坏死因子α可刺激成骨细胞及间充质干细胞中巨噬细胞集落刺激因子的产生,巨噬细胞集落刺激因子通过激活破骨前体细胞中PI3K/Akt、ERK MAPKs通路促进破骨分化,是破骨细胞分化和存活的重要因素[30-32]。 巨噬细胞集落刺激因子与前破骨细胞表面的集落刺激因子1受体结合,刺激RANK的表达。肿瘤坏死因子α还可刺激间充质干细胞释放白细胞介素7,白细胞介素7可激活T细胞产生巨噬细胞集落刺激因子[33-34]。 2.3.5 肿瘤坏死因子α参与不依赖RANK/RANKL的破骨细胞分化途径 部分研究发现,在没有RANK/RANKL的情况下,通过添加适当的细胞因子,可以在体外产生具备功能的破骨细胞。KOBAYASHI等[35]发现与巨噬细胞集落刺激因子共培养的骨髓来源巨噬细胞(bone marrow-derived macrophages,BMDM)可在肿瘤坏死因子α诱导下形成抗酒石酸酸性磷酸酶(tartrate resistant acid phosphatase,TRAP) 阳性的巨噬细胞,且这种诱导可被肿瘤坏死因子1型受体/肿瘤坏死因子2型受体抗体阻断,不能被RANKL的可溶性诱饵受体骨保护素和RANK抗体所阻断。当炎性因子白细胞介素1α存在的情况下,肿瘤坏死因子α诱导的破骨细胞可在骨质表面形成吸收坑。KIM等[36]提取了RANK-/RANKL?小鼠的脾细胞,在肿瘤坏死因子α、巨噬细胞集落刺激因子和转化生长因子β 的共同作用下生成了TRAP+单核细胞。继续在培养物内添加白细胞介素1后,细胞具备了吸收骨质的能力。此外,O’BRIEN等[37]发现RANK-小鼠的关节炎模型中仍存在骨侵蚀现象和TRAP+细胞,肿瘤坏死因子α与白细胞介素6共同作用可在不依赖RANK的情况下在体内和体外诱导破骨细胞形成。 文章总结了肿瘤坏死因子α对破骨细胞的调控研究进展,见表2。"
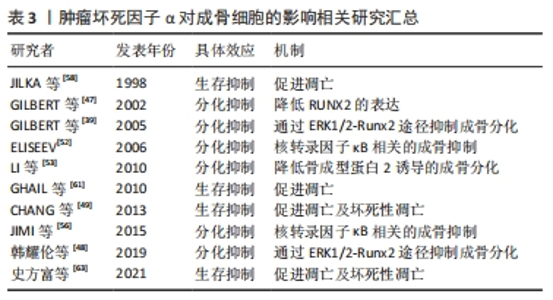
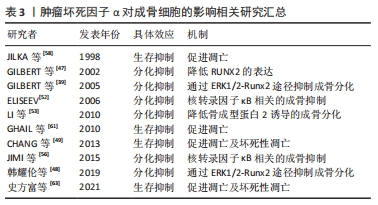
2.4 肿瘤坏死因子α对成骨细胞的影响 肿瘤坏死因子α对成骨细胞的作用概括来看包含2种,生存抑制和分化抑制。通过直接促进处于不同分化阶段的成骨细胞发生凋亡及坏死性凋亡发挥直接杀伤作用,效应的强度受到环境及分化程度的影响,分化程度越高的成骨系细胞效应越显著。同时通过激活多种相关信号通路,抑制成骨相关转录因子,抑制成骨系细胞的成骨分化。 2.4.1 成骨细胞的分化途径与功能 成骨细胞在骨组织的生长改建及骨量维持等方面发挥至关重要的作用。成骨细胞能够产生以Ⅰ型胶原蛋白为主的多种骨基质蛋白。骨基质蛋白以未矿化的类骨质形式分泌后,通过磷酸钙的作用以羟基磷灰石的形式累积矿化。成骨细胞来源于具备多分化潜能的间充质干细胞,通过表达转录因子Runx2和成骨细胞特异性转录因子(Osterix),经骨祖细胞-前成骨细胞转变为成熟的成骨细胞[38]。 2.4.2 肿瘤坏死因子α通过TNFR1/R2调节成骨细胞分化 已知肿瘤坏死因子α显著抑制胎鼠颅顶细胞、MC3T3-E1(clone14)成骨细胞系及人类间充质干细胞的成骨分化。TNFR1是介导肿瘤坏死因子α抑制成骨分化效应的主要受体,因为肿瘤坏死因子1型受体缺陷型间充质干细胞失去了对肿瘤坏死因子α抑制成骨作用的响应。与之相对,野生型或肿瘤坏死因子2型受体缺陷型在与肿瘤坏死因子α共同培养时,出现了矿化结节、碱性磷酸酶及骨钙素表达的不可逆性降低,而肿瘤坏死因子2型受体的存在降低了细胞对肿瘤坏死因子α的敏感性[39]。肿瘤坏死因子1型受体抑制成骨分化的效应可能与其激活核转录因子κB通路抑制转录因子RUNX2和osterix的表达有关,与细胞活力和细胞凋亡无关。 肿瘤坏死因子2型受体在成骨分化中的作用是复杂的,一方面肿瘤坏死因子2型受体可以通过与配体的快速结合/解离提高肿瘤坏死因子1型受体周围肿瘤坏死因子α的浓度,即所谓的“配体浓缩效应”,以此促进肿瘤坏死因子1型受体的表达[40-41]。另一方面,肿瘤坏死因子1型受体和肿瘤坏死因子2型受体作为膜受体也可以通过TACE转化为可溶性形式(sTNFR1和sTNFR2),sTNFR2可与肿瘤坏死因子1型受体竞争性结合肿瘤坏死因子α。此外,在一定的肿瘤坏死因子α浓度范围内,可能存在一种基于TRAF信号传导复合物亲和力的调节机制,肿瘤坏死因子2型受体充当信号抑制器,减弱或改变同一细胞中肿瘤坏死因子1型受体的效应强度[42-44]。 2.4.3 肿瘤坏死因子α通过降低Runx2的表达抑制成骨细胞分化 Runx2是骨形成中必不可少的转录因子,Runx2敲除(Runx2-/-)的小鼠不能形成骨骼。人类Runx2基因单倍体突变会导致锁骨颅骨发育不全综合征(cleidoc-ranial dysplasia,CCD)。RUNX2可直接激活编码骨基质蛋白的基因,如Ⅰ型胶原α1、骨桥蛋白、整合素结合唾液酸蛋白、骨γ-羧基谷氨酸蛋白2和纤连蛋白1[45]。Runx2诱导其他下游转录因子的表达,例如 Osterix 。Runx2 和 Osterix 的表达使间充质干细胞向骨祖细胞分化,促进碱性磷酸酶和 Ⅰ 型胶原蛋白分泌,最终使细胞转变成熟的成骨细胞。Runx2基因包含2种含有独立引物和启动子(P1,P2)的亚型(MASNS和MRIPV)。较短亚型P2/MRIPV在间充质干细胞的分化早期表达,且表达更具组成性。较长亚型P1/MASNS的表达随间充质干细胞向成骨细胞转化而逐渐增加[46]。GILBERT等[47]发现肿瘤坏死因子α可抑制胎鼠颅顶细胞和MC3T3-E1(clone 14)细胞中Runx2基因启动子的活性,降低mRNA的转录水平和稳定性。其中MRIPV mRNA表达被抑制大于90%,MASNS mRNA被抑制达50%,这说明肿瘤坏死因子α阻断了间充质干细胞向成骨细胞分化的早期进程。肿瘤坏死因子α 介导的 Runx2 抑制依赖于 核转录因子κB,但对 Runx2下游靶标 Osx 的抑制与 核转录因子κB 无关。此外,肿瘤坏死因子α可能通过影响ERK1/2-Runx2信号通路从而抑制牙周膜干细胞的成骨分化[48]。 2.4.4 肿瘤坏死因子α通过IKK/核转录因子κB及核转录因子κB/SMAD信号通路抑制成骨分化 如前文所述,肿瘤坏死因子α与受体结合后激活IKKs,进而激活核转录因子κB信号通路。核转录因子κB通路通过诱导 Smad泛素化调节因子1和2促进β-连环蛋白的泛素化和降解,显著抑制间充质干细胞的成骨分化[49-51]。ELISEEV等[52]通过构建腺病毒载体过度表达mIκB(磷酸化位点突变的IκBα,与核转录因子κB结合牢固),在人成骨肉瘤细胞Saos2中抑制核转录因子κB的表达。Saos2细胞表达了更高的成骨表型基因,如碱性磷酸酶、Runx2和骨钙素。 骨形态发生蛋白是转化生长因子 β超家族的一个亚群,在生物体内具有广泛功能,是已知最强大的异位骨化诱导剂之一,在体外细胞培养中碱性磷酸酶、甲状旁腺激素受体、Ⅰ型胶原α1和骨钙素的表达,促进包括间充质干细胞在内的多种细胞/细胞系向成骨细胞分化。骨形态发生蛋白与胞膜表面受体结合后磷酸化特异性信号蛋白 Smad1/5/8,磷酸化的 Smad1/5/8与Smad4形成复合物入细胞核,引起下游靶基因的转录。核转录因子κB 的激活通过减弱 Smad1 活性来抑制骨形态发生蛋白2 诱导的成骨细胞分化[53-55],此外,核转录因子κB p65 亚基可以与细胞核中的 Smad1?Smad5 复合物相互作用并破坏其与靶启动子的结合。使用核转录因子κB拮抗剂阻断肿瘤坏死因子α对核转录因子κB 的激活,可阻止肿瘤坏死因子α抑制 骨形态发生蛋白2诱导的Runx2 mRNA 表达和MC3T3?E1 成骨分化的效应[56-57]。结合前文内容可以发现,核转录因子κB信号通路在肿瘤坏死因子α介导的抗成骨分化作用中处于中心地位,通过阻断核转录因子κB 通路能够抑制肿瘤坏死因子α的抗成骨分化效应,见图3。 2.4.5 肿瘤坏死因子α促进成骨细胞发生凋亡和坏死性凋亡 肿瘤坏死因子α的促凋亡效应与成骨细胞所处环境及成骨细胞的分化成熟程度密切相关,例如低血清环境下肿瘤坏死因子α的促凋亡效应更加明显[58-60]。肿瘤坏死因子α对成骨细胞凋亡的影响也取决于细胞中的Runx2水平。肿瘤坏死因子α通过肿瘤坏死因子1型受体抑制低Runx2表达的骨祖细胞增殖,促进高Runx2表达的成熟成骨细胞凋亡,差异原因可能是低表达的Runx2下调了细胞凋亡过程中的中枢调节因子Bax的表达,缓解了肿瘤坏死因子α诱导的细胞凋亡[61-62]。此外,有研究提到肿瘤坏死因子α增加了人成骨细胞表面Fas的表达,显著增强Fas介导的成骨细胞凋亡。而通过药物增强成骨细胞的自噬可能对凋亡具有一定缓解作用[63-64]。当肿瘤坏死因子α诱导的细胞凋亡被药物抑制时,肿瘤坏死因子α则会诱导成骨细胞发生坏死性凋亡[65-66]。 文章总结了肿瘤坏死因子α对成骨细胞的调控研究进展,见表3。"

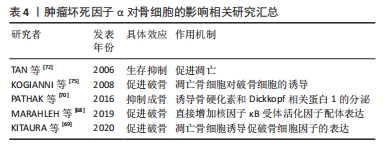
2.5 肿瘤坏死因子α对骨细胞的影响 既往对于骨细胞的研究由于缺乏广泛认可的细胞系及相对苛刻的培养条件而面临较多困难。但由于骨细胞在骨组织中的绝对多数及重要的生理功能,使肿瘤坏死因子α对骨细胞的调控作用难以被忽略。在所有已知的炎性细胞因子中,肿瘤坏死因子α对骨细胞的效应是显著的,包括诱导其凋亡引起破骨促进效应,或通过促进骨细胞表达相关细胞因子发挥间接调控效应。 可预见肿瘤坏死因子α对骨细胞的调控作用仍需在后续的研究中不断探索。 2.5.1 骨细胞的的分化途径与功能 骨细胞是骨组织中最丰富的细胞类型(90%-95%),来源于经历成骨细胞分化的间充质干细胞,外观呈星形/树突状形态。骨基质中的骨细胞通过胞质凸起与相邻的骨细胞构成微小而密集的管隙系统,促进小信号分子,如前列腺素和一氧化氮等炎性因子在细胞间转运传播,从而使骨细胞在炎症调节中发挥重要作用。同样,得益于庞大的数量和表面积,管隙系统内流动的间质液使骨细胞可以感受并传递外界的机械压力,使骨细胞具有机械力传感器的功能。此外,骨细胞还承担至关重要的内分泌及调节其他骨组织细胞生理活动的功能[67]。 2.5.2 肿瘤坏死因子α增强骨细胞中RANKL的表达促进破骨 肿瘤坏死因子α可通过诱导骨细胞中RANKL mRNA的上调促进破骨。将肿瘤坏死因子α处理的骨细胞和肿瘤坏死因子1型受体/肿瘤坏死因子2型受体缺陷的破骨前体细胞共培养,可导致破骨细胞数量显着增加,且此现象可在添加骨保护素后消失,而没有经过肿瘤坏死因子α处理的骨-破骨前体细胞共培养未出现破骨细胞。证明肿瘤坏死因子α通过增强骨细胞中RANKL的表达促进破骨细胞生成,而这一效应是通过激活ERK/JNK/p38 MAPKs和核转录因子κB信号通路实现的[68]。值得一提的是,尽管RANKL可被多种类型细胞表达,但有部分研究指出,骨细胞分泌的RANKL与生理性骨吸收的关系最为密切[69]。 2.5.3 肿瘤坏死因子α增强骨细胞中SOST和DKK1的表达抑制成骨 肿瘤坏死因子α可诱导骨细胞分泌骨硬化素和Dickkopf相关蛋白1。骨硬化素和Dickkopf相关蛋白1 通过抑制Wnt /β-catenin通路对成骨细胞分化和活性的促进作用来减少骨形成,导致广泛性骨质流失,而抑制核转录因子κB信号传导可抑制骨硬化素的分泌[70-71]。 2.5.4 肿瘤坏死因子α增强骨细胞的凋亡及凋亡骨细胞诱导的骨吸收 近年来,肿瘤坏死因子α诱导骨细胞凋亡的现象在众多研究中被提及,其与地塞米松及依托泊苷等常在研究中被用来诱导骨细胞凋亡。成骨细胞和骨细胞等成熟分化的细胞,比骨膜成纤维细胞和周围的骨祖细胞等未成熟细胞,对肿瘤坏死因子α诱导的凋亡更加敏感。施加外界应力可以显著抑制肿瘤坏死因子α诱导的骨细胞凋亡,可能由于骨细胞在应力作用下迅速释放少量一氧化氮抑制了人半胱氨酸蛋白酶3的活性[72-73]。这表明骨细胞凋亡在调节骨重塑过程中的破骨细胞骨吸收效应中起重要作用,尤其是在应力负荷条件下。但有研究提及在无血清培养环境下,肿瘤坏死因子α失去了对小鼠骨样细胞(MLO-Y4)中与凋亡相关半胱天冬酶3/7活性的影响[74],说明肿瘤坏死因子α的促凋亡效应可能与细胞所处环境相关。 已凋亡的骨细胞可以刺激破骨细胞的生成。KOGIANNI等[75]发现骨细胞凋亡体可诱发破骨细胞生成和骨吸收反应,此类反应的出现不依赖于RANKL,而在体外和体内添加肿瘤坏死因子α中和抗体可显著阻断这一反应,因此抑制骨细胞的凋亡被认为具有治疗骨质丢失/疏松的潜在价值。双膦酸盐和降钙素类药物,以及肿瘤坏死因子受体/配体超家族成员CD40/CD40L,都被报道显著抑制了肿瘤坏死因子α诱导的MLO-Y4细胞的凋亡[76-77]。同时,凋亡的骨细胞促进周围的骨细胞和巨噬细胞分泌多种炎性及生长因子,如 RANKL、肿瘤坏死因子α、白细胞介素1β、白细胞介素6、白细胞介素8和血管内皮生长因子,这些细胞因子均被认为促进了破骨细胞的生成[69],见图4。"

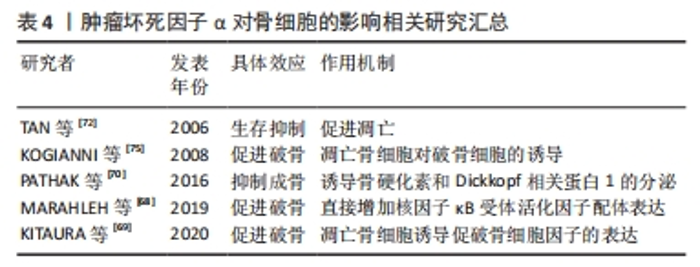
文章总结了肿瘤坏死因子α对骨细胞的调控相关研究进展,见表4。"
| [1] ANSARI N, SIMS NA. The cells of bone and their interactions. Handb Exp Pharmacol. 2020;262:1-25. [2] KONDO N, KURODA T, KOBAYASHI D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci. 2021;22(20):10922. [3] JANG D, LEE A, SHIN H, et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci. 2021;22(5):2719. [4] KITAURA H, MARAHLEH A, OHORI F, et al. Role of the interaction of tumor necrosis factor-α and tumor necrosis factor receptors 1 and 2 in bone-related cells. Int J Mol Sci. 2022;23(3):1481. [5] IDRESS M, MILNE BF, THOMPSON GS, et al. Structure-based design, synthesis and bioactivity of a new anti-TNFα cyclopeptide. Molecules. 2020;25(4):922. [6] YONGYUN C, JINGWEI Z, ZHIQING L, et al. Andrographolide stimulates osteoblastogenesis and bone formation by inhibiting nuclear factor kappa-Β signaling both in vivo and in vitro. J Orthop Translat. 2019;19:47-57. [7] DONSANTE S, PALMISANO B, SERAFINI M, et al. From stem cells to bone-forming cells. Int J Mol Sci. 2021;22(8):3989. [8] EVANS CE, JONES S. Soluble factors secreted by macrophage-like cells in vitro cause osteoprogenitor cell detachment. Calcif Tissue Int. 1998,63(6):496-504. [9] GERSTENFELD LC, CHO TJ, KON T, et al. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs. 2001;169(3):285-294. [10] HARBOUR ME, GREGORY JW, JENKINS HR, et al. Proliferative response of different human osteoblast-like cell models to proinflammatory cytokines. Pediatr Res. 2000;48(2):163-168. [11] LEE S, SHARMA AR, CHOI B, et al. The effect of TNFα secreted from macrophages activated by titanium particles on osteogenic activity regulated by WNT/BMP signaling in osteoprogenitor cells. Biomaterials. 2012;33(17):4251-4263. [12] ZHANG L, HADDOUTI EM, WELLE K, et al. The effects of biomaterial implant wear debris on osteoblasts. Front Cell Dev Biol. 2020;8:352. [13] BEN DAVID D, REZNICK AZ, SROUJI S, et al. Exposure to pro-inflammatory cytokines upregulates MMP-9 synthesis by mesenchymal stem cells-derived osteoprogenitors. Histochem Cell Biol. 2008;129(5):589-597. [14] LIU H, LI D, ZHANG Y, et al. Inflammation, mesenchymal stem cells and bone regeneration. Histochem Cell Biol. 2018;149(4):393-404. [15] BEN-DAVID D, LIVNE E, REZNICK AZ. The involvement of oxidants and NF-kappaB in cytokine-induced MMP-9 synthesis by bone marrow-derived osteoprogenitor cells. Inflamm Res. 2012;61(7):673-688. [16] GUO H, ZHANG J, HAO S, et al. Adenovirus-mediated small interfering RNA targeting tumor necrosis factor-α inhibits titanium particle-induced osteoclastogenesis and bone resorption. Int J Mol Med. 2013;32(2):296-306. [17] SUN Y, LI J, XIE X, et al. Macrophage-osteoclast associations: origin, polarization, and subgroups. Front Immunol. 2021;12:778078. [18] BORGHI A, VERSTREPEN L, BEYAERT R. TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem Pharmacol. 2016;116:1-10. [19] YAO Z, GETTING SJ, LOCKE IC. Regulation of TNF-induced osteoclast differentiation. Cells. 2021;11(1):132. [20] PARK H, BAEK K, BAEK J, et al. TNFα increases RANKL expression via PGE2-induced activation of NFATc1. Int J Mol Sci. 2017;18(3):495. [21] YOU K, GU H, YUAN Z, et al. Tumor necrosis factor alpha signaling and organogenesis. Front Cell Dev Biol. 2021;9:727075. [22] MUKAI T, OTSUKA F, OTANI H, et al. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem Biophys Res Commun. 2007;356(4):1004-1010. [23] ISLAM S, HASSAN F, TUMURKHUU G, et al. Bacterial lipopolysaccharide induces osteoclast formation in RAW 264.7 macrophage cells. Biochem Biophys Res Commun. 2007;360(2):346-351. [24] SADEK KM, EL MS, RADWAN IA, et al. Molecular basis beyond interrelated bone resorption/regeneration in periodontal diseases: a concise review. Int J Mol Sci. 2023;24(5):4599. [25] ALFORD PB, XUE Y, SHACKELFORD RE. Tumor necrosis factor-alpha induces c-fms RNA expression in murine tissue macrophages. Biochem Biophys Res Commun. 1997;240(2):442-445. [26] KITAURA H, SANDS MS, AYA K, et al. Marrow stromal cells and osteoclast precursors differentially contribute to TNF-α-induced osteoclastogenesis in vivo. J Immunol. 2004;173(8):4838. [27] MARAHLEH A, KITAURA H, OHORI F, et al. Effect of TNF-α on osteocyte RANKL expression during orthodontic tooth movement. J Dent Sci. 2021;16(4):1191-1197. [28] OGAWA S, KITAURA H, KISHIKAWA A, et al. TNF-α is responsible for the contribution of stromal cells to osteoclast and odontoclast formation during orthodontic tooth movement. PLoS One. 2019;14(10):e223989. [29] ZOU W, AMCHESLAVSKY A, TAKESHITA S, et al. TNF-α expression is transcriptionally regulated by RANK ligand. J Cell Physiol. 2005;202(2):371-378. [30] KITAURA H, ZHOU P, KIM H, et al. M-CSF mediates TNF-induced inflammatory osteolysis. J Clin Invest. 2005;115(12):3418-3427. [31] HU X, YIN Z, CHEN X, et al. Tussilagone inhibits osteoclastogenesis and periprosthetic osteolysis by suppressing the NF-κB and P38 MAPK signaling pathways. Front Pharmacol. 2020;11:385. [32] STARLINGER J, SARAHRUDI K, KECHT M, et al. The influence of M-CSF on fracture healing in a mouse model. Sci Rep. 2021;11(1):22326. [33] MEYER A, PARMAR PJ, SHAHRARA S. Significance of IL-7 and IL-7R in RA and autoimmunity. Autoimmun Rev. 2022;21(7):103120. [34] KIM JH, SIM JH, LEE S, et al. Interleukin-7 induces osteoclast formation via STAT5, independent of receptor activator of NF-kappaB ligand. Front Immunol. 2017;8:1376. [35] KOBAYASHI K, TAKAHASHI N, JIMI E, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191(2):275-286. [36] KIM N, KADONO Y, TAKAMI M, et al. Osteoclast differentiation independent of the TRANCE–RANK–TRAF6 axis. J Exp Med. 2005;202(5):589-595. [37] O’BRIEN W, FISSEL BM, MAEDA Y, et al. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 2016;68(12): 2889-2900. [38] MIZOGUCHI T, ONO N. The diverse origin of bone-forming osteoblasts. J Bone Miner Res. 2021;36(8):1432-1447. [39] GILBERT LC, RUBIN J, NANES MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab. 2005;288(5):E1011-E1018. [40] TARTAGLIA LA, PENNICA D, GOEDDEL D . Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268(25):18542-18548. [41] SETHI JK, HOTAMISLIGIL GS. Metabolic messengers: tumour necrosis factor. Nat Metab. 2021;3(10):1302-1312. [42] BRENNER D, BLASER H, MAK TW. Regulation of tumour necrosis factor signalling:live or let die. Nat Rev Immunol. 2015;15(6):362-374. [43] PESCATORE A, CASALE C, FUSCO F, et al. Methods to study the effect of IKK inhibition on TNF-inducing apoptosis and necroptosis in cultured cells. Methods Mol Biol. 2021;2366:243-254. [44] MCMILLAN D, MARTINEZ-FLEITES C, PORTER J, et al. Structural insights into the disruption of TNF-TNFR1 signalling by small molecules stabilising a distorted TNF. Nat Commun. 2021;12(1):582. [45] KOMORI T. Regulation of proliferation, differentiation and functions of osteoblasts by Runx2. Int J Mol Sci. 2019;20(7):1694. [46] KIM H, KIM W, RYOO H. Post-translational regulations of transcriptional activity of RUNX2. Molecules and Cells. 2020;43(2):160-167. [47] GILBERT L, HE X, FARMER P, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2αA) is inhibited by tumor necrosis factor-α. J Biol Chem. 2002;277(4):2695-2701. [48] 韩耀伦,王璐,马欣.ERK1/2-Runx2信号通路在TNF-α对牙周膜干细胞成骨分化影响中的作用[J].中国免疫学杂志,2019;35(21):2587-2592. [49] CHANG J, LIU F, LEE M, et al. NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation. Proc Natl Acad Sci U S A. 2013;110(23):9469-9474. [50] LI W, LIU Q, SHI J, et al. The role of TNF-α in the fate regulation and functional reprogramming of mesenchymal stem cells in an inflammatory microenvironment. Front Immunol. 2023;14:1074863. [51] CHOU LY, HO CT, HUNG SC. Paracrine senescence of mesenchymal stromal cells involves inflammatory cytokines and the NF-κB pathway. Cells. 2022;11(20):3324. [52] ELISEEV R, SCHWARZ E, ZUSCIK M, et al. Smad7 mediates inhibition of Saos2 osteosarcoma cell differentiation by NFκB. Exp Cell Res. 2006;312(1):40-50. [53] LI W, YU B, LI M, et al. NEMO-binding domain peptide promotes osteoblast differentiation impaired by tumor necrosis factor alpha. Biochem Biophys Res Commun. 2010;391(2):1228-1233. [54] LI X, YANG H, ZHANG Z, et al. Concentrated growth factor exudate enhances the proliferation of human periodontal ligament cells in the presence of TNF‑α. Mol Med Rep. 2019;19(2):943-950. [55] WU WJ, XIA CL, OU SJ, et al. Prophylactic effects of NFκB essential modulator-binding domain peptides on bone infection:an experimental study in a rabbit model. J Inflamm Res. 2022;15:2745-2759. [56] JIMI E. The role of BMP signaling and NF-kappaB signaling on osteoblastic differentiation, cancer development, and vascular diseases--is the activation of NF-kappaB a friend or foe of BMP function? Vitam Horm. 2015;99:145-170. [57] ZHU WQ, MING PP, ZHANG SM, et al. Role of MAPK/JNK signaling pathway on the regulation of biological behaviors of MC3T3‑E1 osteoblasts under titanium ion exposure. Mol Med Rep. 2020;22(6):4792-4800. [58] JILKA RL, WEINSTEIN RS, BELLIDO T, et al. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res. 1998;13(5):793-802. [59] HAMEISTER R, LOHMANN CH, DHEEN ST, et al. The effect of TNF-α on osteoblasts in metal wear-induced periprosthetic bone loss. Bone Joint Res. 2020;9(11):827-839. [60] LUTTRELL LM, DAR MS, GESTY-PALMER D, et al. Transcriptomic characterization of signaling pathways associated with osteoblastic differentiation of MC-3T3E1 cells. PLoS One. 2019;14(1):e204197. [61] GHALI O, CHAUVEAU C, HARDOUIN P, et al. TNF-α’s effects on proliferation and apoptosis in human mesenchymal stem cells depend on RUNX2 expression. J Bone and Miner Res. 2010;25(7):1616-1626. [62] KOMORI T. Molecular mechanism of Runx2-dependent bone development. Mol Cells. 2020;43(2):168-175. [63] 史方富,崔红旺,孙博.自噬在肿瘤坏死因子α诱导成骨细胞凋亡中的作用[J].临床骨科杂志,2021,24(4):601-604. [64] XIAO L, XIAO Y. The Autophagy in osteoimmonology: self-eating, maintenance, and beyond. Front Endocrinol (Lausanne). 2019;10:490. [65] SHI G, JIA P, CHEN H, et al. Necroptosis occurs in osteoblasts during tumor necrosis factor-α stimulation and caspase-8 inhibition. Braz J Med Biol Res. 2019; 52(1):e7844. [66] HU X, WANG Z, KONG C, et al. Necroptosis: a new target for prevention of osteoporosis. Front Endocrinol (Lausanne). 2022;13:1032614. [67] TRESGUERRES FGF, TORRES J, LÓPEZ-QUILES J, et al. The osteocyte:a multifunctional cell within the bone. Ann Anat. 2020;227:151422. [68] MARAHLEH A, KITAURA H, OHORI F, et al. TNF-α directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front Immunol. 2019; 10:2925. [69] KITAURA H, MARAHLEH A, OHORI F, et al. Osteocyte-related cytokines regulate osteoclast formation and bone resorption. Int J Mol Sci. 2020;21(14):5169. [70] PATHAK JL, BAKKER AD, LUYTEN FP, et al. Systemic inflammation affects human osteocyte-specific protein and cytokine expression. Calcif Tissue Int. 2016;98(6):596-608. [71] RAUNER M, TAIPALEENMÄKI H, TSOURDI E, et al. Osteoporosis treatment with anti-sclerostin antibodies-mechanisms of action and clinical application. J Clin Med. 2021;10(4):787. [72] TAN SD, KUIJPERS-JAGTMAN AM, SEMEINS CM, et al. Fluid shear stress inhibits tnfα-induced osteocyte apoptosis. J Dental Res. 2006;85(10):905-909. [73] YAN Y, WANG L, GE L, et al. Osteocyte-mediated translation of mechanical stimuli to cellular signaling and its role in bone and non-bone-related clinical complications. Curr Osteoporos Rep. 2020;18(1):67-80. [74] BAKKER AD, DA SILVA VC, KRISHNAN R, et al. Tumor necrosis factor α and interleukin-1β modulate calcium and nitric oxide signaling in mechanically stimulated osteocytes. Arthritis Rheum. 2009;60(11):3336-3345. [75] KOGIANNI G, MANN V, NOBLE BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23(6):915-927. [76] PLOTKIN LI, WEINSTEIN RS, PARFITT AM, et al. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999; 104(10):1363-1374. [77] AHUJA SS, ZHAO S, BELLIDO T, et al. CD40 ligand blocks apoptosis induced by tumor necrosis factor α, glucocorticoids, and etoposide in osteoblasts and the osteocyte-like cell line murine long bone osteocyte-Y4. Endocrinology. 2003; 144(5):1761-1769. |
| [1] | Yang Yufang, Yang Zhishan, Duan Mianmian, Liu Yiheng, Tang Zhenglong, Wang Yu. Application and prospects of erythropoietin in bone tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(9): 1443-1449. |
| [2] | Chen Kaijia, Liu Jingyun, Cao Ning, Sun Jianbo, Zhou Yan, Mei Jianguo, Ren Qiang. Application and prospect of tissue engineering in treatment of osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(9): 1450-1456. |
| [3] | Yang Yifeng, Ye Nan, Wang Lin, Guo Shuaicheng, Huang Jian. Signaling pathway of dexmedetomidine against ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(9): 1464-1469. |
| [4] | Huang Haoran, Fan Yinuo, Wei-Yang Wenxiang, Jiang Mengyu, Fang Hanjun, Wang Haibin, Chen Zhenqiu, Liu Yuhao, Zhou Chi. Urolithin A mediates p38/MAPK pathway to inhibit osteoclast activity [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1149-1154. |
| [5] | Yue Yun, Wang Peipei, Yuan Zhaohe, He Shengcun, Jia Xusheng, Liu Qian, Li Zhantao, Fu Huiling, Song Fei, Jia Menghui. Effects of croton cream on JNK/p38 MAPK signaling pathway and neuronal apoptosis in cerebral ischemia-reperfusion injury rats [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1186-1192. |
| [6] | Liu Xin, Hu Man, Zhao Wenjie, Zhang Yu, Meng Bo, Yang Sheng, Peng Qing, Zhang Liang, Wang Jingcheng. Cadmium promotes senescence of annulus fibrosus cells via activation of PI3K/Akt signaling pathway [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1217-1222. |
| [7] | Wei Juan, Li Ting, Huan Mengting, Xie Ying, Xie Zhouyu, Wei Qingbo, Wu Yunchuan. Mechanism by which static exercise improves insulin resistance in skeletal muscle of type 2 diabetes [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1271-1276. |
| [8] | Wang Wen, Zheng Pengpeng, Meng Haohao, Liu Hao, Yuan Changyong. Overexpression of Sema3A promotes osteogenic differentiation of dental pulp stem cells and MC3T3-E1 [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 993-999. |
| [9] | Liu Jianhong, Liao Shijie, Li Boxiang, Tang Shengping, Wei Zhendi, Ding Xiaofei. Extracellular vesicles carrying non-coding RNA regulate the activation of osteoclasts [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 1076-1082. |
| [10] | Wang Shanshan, Shu Qing, Tian Jun. Physical factors promote osteogenic differentiation of stem cells [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 1083-1090. |
| [11] | Zhang Kefan, Shi Hui. Research status and application prospect of cytokine therapy for osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(6): 961-967. |
| [12] | Wang Jiani, Chen Junyu. Angiogenesis mechanism of metal ions and their application in bone tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(5): 804-812. |
| [13] | Zhu Liwei, Wang Jiangyue, Bai Ding. Application value of nanocomposite gelatin methacryloyl hydrogels in different bone defect environments [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(5): 753-758. |
| [14] | Wei Yuanxun, Chen Feng, Lin Zonghan, Zhang Chi, Pan Chengzhen, Wei Zongbo. The mechanism of Notch signaling pathway in osteoporosis and its prevention and treatment with traditional Chinese medicine [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(4): 587-593. |
| [15] | Shen Feiyan, Yao Jixiang, Su Shanshan, Zhao Zhongmin, Tang Weidong. Knockdown of circRNA WD repeat containing protein 1 inhibits proliferation and induces apoptosis of chondrocytes in knee osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(4): 499-504. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||