Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (34): 8978-8985.doi: 10.12307/2026.771
Previous Articles Next Articles
Lentivirus-mediated gene therapy in a beta-thalassemia mouse model
Liu Hongwei, Chang Lungji
- School of Medicine, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan Province, China
-
Received:2025-09-29Revised:2026-01-13Online:2026-12-08Published:2026-04-14 -
Contact:Chang Lungji, PhD, Professor, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan Province, China -
About author:Liu Hongwei, PhD candidate, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan Province, China
CLC Number:
Cite this article
Liu Hongwei, Chang Lungji. Lentivirus-mediated gene therapy in a beta-thalassemia mouse model [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(34): 8978-8985.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
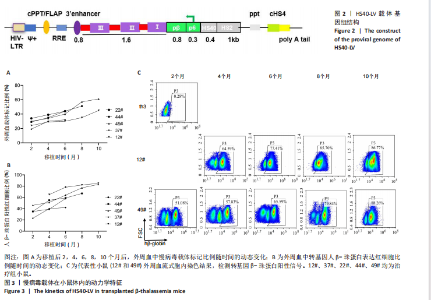
2.1 慢病毒载体 构建前病毒DNA载体HS40-LV,见图2。该载体携带突变型HBB基因,由上游的pβ/p6/HS40/HS2调控元件驱动。通过病毒包装产生高滴度的病毒液,滴度约为10×108 TU/mL。 2.2 实验动物数量分析 治疗组小鼠中途死亡1只,数据丢失,剩余小鼠进入结果分析。 2.3 慢病毒载体体内动力学特征 在移植后2,4,6,8,10个月,通过qPCR方法检测到5只治疗小鼠(12#、22#、37#、44#、49#)外周血中载体标记比例随时间持续增加,第10个月时5只小鼠的平均载体标记比例为42%,49#小鼠达到最高(60%),见图3A。每只小鼠血液中人β-珠蛋白阳性红细胞比例均逐渐增加,在移植后10个月,5只小鼠的人β-珠蛋白阳性红细胞平均比例为70%,最高达到80%(12#和49#小鼠),见图3B,C。中等载体标记的小鼠实现了血液中人β-珠蛋白阳性红细胞的高水平,例如,治疗4个月的小鼠平均载体标记低于30%,有超过50%的人β-珠蛋白阳性红细胞;在6个月时有40%的载体标记,有60%的人β-珠蛋白阳性红细胞;在8个月时有50%"
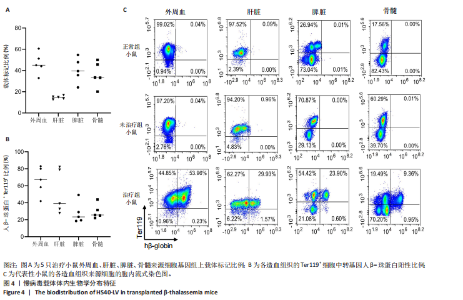
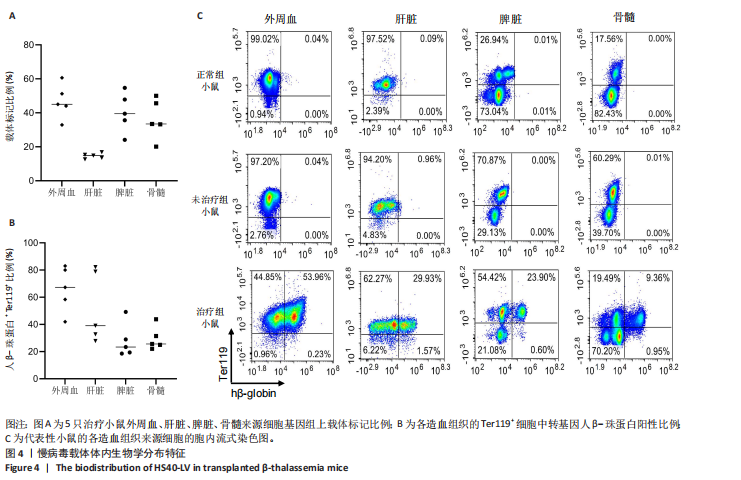
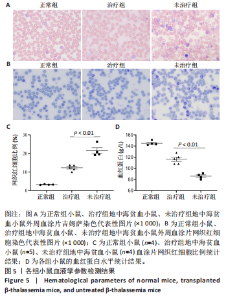
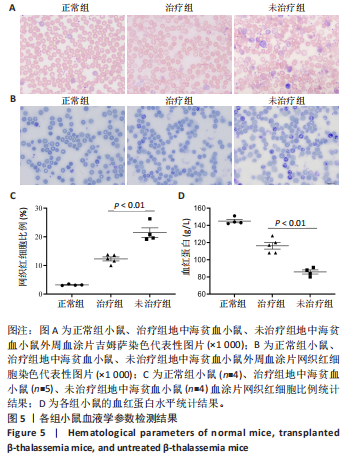
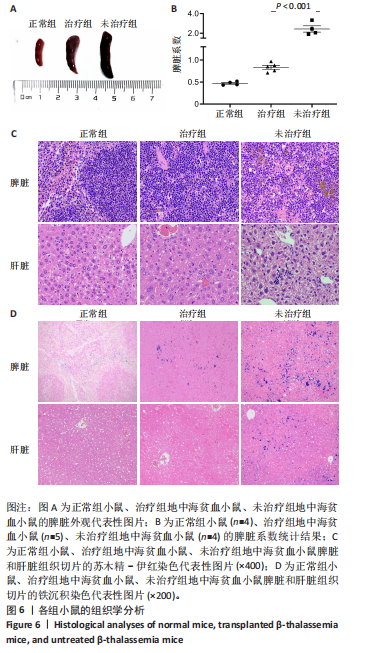
的载体标记,约70%的人β-珠蛋白阳性红细胞。整个随访期间,未见人β-珠蛋白阳性红细胞水平下降,表明转基因在体内没有出现表达沉默的现象。 2.4 载体体内生物学分布 在移植后10个月,通过qPCR检测到5只治疗小鼠外周血DNA样本中载体标记比例为(46.74±4.55)%,肝脏中为(14.59±0.72)% ,脾脏中为(40.34±5.23)%,骨髓中为(36.48±5.27)%,见图4A。通过流式胞内染色,分别在血液、肝脏、脾脏和骨髓来源细胞样本中检测到表达人β-珠蛋白的Ter119+细胞比例为(66.14±7.49)%,(40.21±10.98)%,(21.60±2.08)%和(24.79±1.96)%(图4B)。人β-珠蛋白阳性信号只出现在Ter119+细胞(小鼠红细胞)群中,未出现在Ter119-细胞群中,见图4C,表明转基因人β-珠蛋白具有高度的红系特异性。 2.5 功能矫正 2.5.1 血液学参数改善 在未治疗的th3地中海贫血小鼠外周血涂片中,异形性细胞和细胞碎片较多,染色深浅不一,见图5A。治疗10个月后,小鼠异形性红细胞和小细胞显著减少,形态和染色深浅较均一,但仍有少量异常细胞,见图5A。在未治疗组th3小鼠的血液中网织红细胞(着色为深蓝色)的平均比例为22.07%,治疗组小鼠的网织红细胞数量显著减少至12.66%(P < 0.01),见图5B,C。正常组小鼠中网织红细胞比例很低,见图5B,C。由于缺乏b1和b2 基因,th3小鼠产生的β-珠蛋白量降低,血红蛋白水平低至(84.75±1.79) g/L,表现为贫血。治疗后的th3小鼠的平均血红蛋白水平为(116.5±3.23) g/L,相较于th3小鼠,提高了约37%(P < 0.01),见图5D。 2.5.2 组织病理学改善 在地中海贫血小鼠中,脾脏肿大明显。正常小鼠脾脏体积较小、颜色鲜红。与未治疗组小鼠相比,治疗组小鼠脾脏大小有所改善,见图6A。治疗组小鼠脾脏系数显著低于未治疗组(P < 0.001),见图6B。苏木精-伊红染色显示未治疗组小鼠脾脏红髓和白髓之间的边界紊乱,且红髓区域扩大,见图6C;治疗组小鼠脾脏中红髓区域逐渐减少,显示出脾脏结构和造血功能的改善,见图6C。在未治疗组小鼠中,肝脏中有少量染色为深蓝的单个核细胞簇,该类细胞为红细胞前体,红系祖细胞浸润显示轻度髓外造血,见图6C;治疗组小鼠单个核细胞的残余极少,表明造血压力得到缓解。在未治疗组小鼠脾脏中检测到大量的铁沉积,见图6D,显示脾脏是主要受累器官;治疗组小鼠脾脏中铁沉积水平显著下降,见图6D。在地中海贫血小鼠中,肝脏组织的内源性铁沉积增加,但是沉积水平较脾脏低;治疗组和正常组小鼠的肝脏铁沉积水平很低,见图6D。"
"

"
| [1] KHANDROS E, THOM CS, D’SOUZA J, et al. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood. 2012;119(22): 5265-5275. [2] SANCHEZ-VILLALOBOS M, BLANQUER M, MORALEDA JM, et al. New Insights Into Pathophysiology of β-Thalassemia. Front Med (Lausanne). 2022;9:880752. [3] FARMAKIS D, PORTER J, TAHER A, et al. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. Hemasphere. 2022;6(8):e732. [4] TAHER AT, MUSALLAM KM, CAPPELLINI MD. β-Thalassemias. N Engl J Med. 2021; 384(8):727-743. [5] ALGERI M, LODI M, LOCATELLI F. Hematopoietic Stem Cell Transplantation in Thalassemia. Hematol Oncol Clin North Am. 2023;37(2):413-432. [6] LUCARELLI G, GAZIEV J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22(2):53-63. [7] BARONCIANI D, ANGELUCCI E, POTSCHGER U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant. 2016;51(4):536-541. [8] PSATHA N, PAPAYANNI PG, YANNAKI E. A New Era for Hemoglobinopathies: More Than One Curative Option. Curr Gene Ther. 2017;17(5):364-378. [9] LEONARD A, TISDALE JF, BONNER M. Gene Therapy for Hemoglobinopathies: Beta-Thalassemia, Sickle Cell Disease. Hematol Oncol Clin North Am. 2022;36(4): 769-795. [10] FERRARI G, CAVAZZANA M, MAVILIO F. Gene Therapy Approaches to Hemoglobinopathies. Hematol Oncol Clin North Am. 2017;31(5):835-852. [11] MAGRIN E, SEMERARO M, HEBERT N, et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: the HGB-205 trial. Nat Med. 2022; 28(1):81-88. [12] KWIATKOWSKI JL, WALTERS MC, HONGENG S, et al. Betibeglogene autotemcel gene therapy in patients with transfusion-dependent, severe genotype β-thalassaemia (HGB-212): a non-randomised, multicentre, single-arm, open-label, single-dose, phase 3 trial. Lancet. 2024;404(10468):2175-2186. [13] MIRZA A, RITSERT ML, TAO G, et al. Gene therapy in transfusion-dependent non-β0/β0 genotype β-thalassemia: first real-world experience of beti-cel. Blood Adv. 2025;9(1):29-38. [14] THOMPSON AA, WALTERS MC, KWIATKOWSKI J, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2018;378(16):1479-1493. [15] MARKTEL S, SCARAMUZZA S, CICALESE MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. 2019;25(2):234-241. [16] ROSELLI EA, MEZZADRA R, FRITTOLI MC, et al. Correction of beta-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol Med. 2010;2(8):315-328. [17] BOULAD F, MAGGIO A, WANG X, et al. Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: a phase 1 trial. Nat Med. 2022;28(1):63-70. [18] OUYANG W, DONG G, ZHAO W, et al. Restoration of β-Globin Expression with Optimally Designed Lentiviral Vector for β-Thalassemia Treatment in Chinese Patients. Hum Gene Ther. 2021;32(9-10):481-494. [19] LI S, LING S, WANG D, et al. Modified lentiviral globin gene therapy for pediatric β(0)/β(0) transfusion-dependent β-thalassemia: A single-center, single-arm pilot trial. Cell Stem Cell. 2024;31(7):961-973. [20] HARRISON C. First gene therapy for β-thalassemia approved. Nature Biotechnology. 2019;37(10):1102-1103. [21] JONES RJ, DEBAUN MR. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood. 2021;138(11):942-947. [22] DUNCAN CN, BLEDSOE JR, GRZYWACZ B, et al. Hematologic Cancer after Gene Therapy for Cerebral Adrenoleukodystrophy. N Engl J Med. 2024;391(14):1287-1301. [23] ROMERO Z, URBINATI F, GEIGER S, et al. β-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest. 2013;123(8):3317-3330. [24] NEGRE O, EGGIMANN AV, BEUZARD Y, et al. Gene Therapy of the β-Hemoglobinopathies by Lentiviral Transfer of the β(A(T87Q))-Globin Gene. Hum Gene Ther. 2016;27(2):148-165. [25] MORGAN RA, UNTI MJ, ALESHE B, et al. Improved Titer and Gene Transfer by Lentiviral Vectors Using Novel, Small β-Globin Locus Control Region Elements. Mol Ther. 2020;28(1):328-340. [26] HANAWA H, HARGROVE PW, KEPES S, et al. Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood. 2004;104(8):2281-2290. [27] LISOWSKI L, SADELAIN M. Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood. 2007;110(13):4175-4178. [28] MICCIO A, CESARI R, LOTTI F, et al. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(30):10547-10552. [29] RIVELLA S, SADELAIN M. Genetic treatment of severe hemoglobinopathies: the combat against transgene variegation and transgene silencing. Semin Hematol. 1998;35(2):112-125. [30] YANNAKI E, TUBB J, AKER M, et al. Topological constraints governing the use of the chicken HS4 chromatin insulator in oncoretrovirus vectors. Mol Ther. 2002; 5(5 Pt 1):589-598. [31] KAISER J. Gene therapy. Seeking the cause of induced leukemias in X-SCID trial. Science. 2003;299(5606):495. [32] EMERY DW, YANNAKI E, TUBB J, et al. Development of virus vectors for gene therapy of beta chain hemoglobinopathies: flanking with a chromatin insulator reduces gamma-globin gene silencing in vivo. Blood. 2002;100(6):2012-2019. [33] BROWNING DL, TROBRIDGE GD. Insulators to Improve the Safety of Retroviral Vectors for HIV Gene Therapy. Biomedicines. 2016;4(1):4. [34] GOODMAN MA, ARUMUGAM P, PILLIS DM, et al. Foamy Virus Vector Carries a Strong Insulator in Its Long Terminal Repeat Which Reduces Its Genotoxic Potential. J Virol. 2018;92(1):e01639-17. [35] IMREN S, PAYEN E, WESTERMAN KA, et al. Permanent and panerythroid correction of murine beta thalassemia by multiple lentiviral integration in hematopoietic stem cells. Proc Natl Acad Sci U S A. 2002;99(22):14380-14385. [36] MAY C, RIVELLA S, CHADBURN A, et al. Successful treatment of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Blood. 2002;99(6):1902-1908. [37] NEGRE O, FUSIL F, COLOMB C, et al. Correction of murine β-thalassemia after minimal lentiviral gene transfer and homeostatic in vivo erythroid expansion. Blood. 2011;117(20):5321-5331. [38] PERSONS DA, ALLAY ER, SABATINO DE, et al. Functional requirements for phenotypic correction of murine beta-thalassemia: implications for human gene therapy. Blood. 2001;97(10):3275-3282. [39] ANDREANI M, NESCI S, LUCARELLI G, et al. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. Bone Marrow Transplant. 2000;25(4):401-404. [40] FITZHUGH CD, CORDES S, TAYLOR T, et al. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017; 130(17):1946-1948. [41] AIUTI A, SLAVIN S, AKER M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002; 296(5577):2410-2413. [42] RAFTOPOULOS H, WARD M, LEBOULCH P, et al. Long-term transfer and expression of the human beta-globin gene in a mouse transplant model. Blood. 1997;90(9):3414-3422. [43] NEGRE O, BARTHOLOMAE C, BEUZARD Y, et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of β-thalassemia and sickle cell disease. Curr Gene Ther. 2015;15(1):64-81. [44] FINKELSHTEIN D, WERMAN A, NOVICK D, et al. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110(18):7306-7311. [45] LEBOULCH P, HUANG GM, HUMPHRIES RK, et al. Mutagenesis of retroviral vectors transducing human beta-globin gene and beta-globin locus control region derivatives results in stable transmission of an active transcriptional structure. EMBO J. 1994;13(13):3065-3076. [46] WANG CX, SATHER BD, WANG X, et al. Rapamycin relieves lentiviral vector transduction resistance in human and mouse hematopoietic stem cells. Blood. 2014;124(6):913-923. [47] DELVILLE M, SOHEILI T, BELLIER F, et al. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol Ther Methods Clin Dev. 2018;10:341-347. [48] PETRILLO C, CALABRIA A, PIRAS F, et al. Assessing the Impact of Cyclosporin A on Lentiviral Transduction and Preservation of Human Hematopoietic Stem Cells in Clinically Relevant Ex Vivo Gene Therapy Settings. Hum Gene Ther. 2019;30(9):1133-1146. [49] MASIUK KE, ZHANG R, OSBORNE K, et al. PGE2 and Poloxamer Synperonic F108 Enhance Transduction of Human HSPCs with a β-Globin Lentiviral Vector. Mol Ther Methods Clin Dev. 2019;13:390-398. [50] UCHIDA N, NASSEHI T, DRYSDALE CM, et al. High-Efficiency Lentiviral Transduction of Human CD34(+) Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol Ther Methods Clin Dev. 2019;13:187-196. [51] LEVASSEUR DN, RYAN TM, PAWLIK KM, et al. Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood. 2003;102(13):4312-4319. [52] RIVELLA S, MAY C, CHADBURN A, et al. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101(8):2932-2939. [53] GOTT KM, POTTER CA, DOYLE-EISELE M, et al. A Comparison of Cs-137 γ Rays and 320-kV X-Rays in a Mouse Bone Marrow Transplantation Model. Dose Response. 2020;18(2):1559325820916572. [54] LI Z, DÜLLMANN J, SCHIEDLMEIER B, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296(5567):497. [55] UCHIDA N, WEITZEL RP, EVANS ME, et al. Evaluation of engraftment and immunological tolerance after reduced intensity conditioning in a rhesus hematopoietic stem cell gene therapy model. Gene Ther. 2014;21(2):148-157. [56] WYSS BK, MEYERS JL, SINN AL, et al. A novel competitive repopulation strategy to quantitate engraftment of ex vivo manipulated murine marrow cells in submyeloablated hosts. Exp Hematol. 2008;36(4):513-521. [57] MARDINEY M 3RD, JACKSON SH, SPRATT SK, et al. Enhanced host defense after gene transfer in the murine p47phox-deficient model of chronic granulomatous disease. Blood. 1997;89(7):2268-2275. [58] COWEN D, RICHAUD P, MARIT G, et al. Regimen-related toxicity in patients undergoing BMT with total body irradiation using a sweeping beam technique. Bone Marrow Transplant. 1992;10(6):515-519. [59] BEARMAN SI, APPELBAUM FR, BACK A, et al. Regimen-related toxicity and early posttransplant survival in patients undergoing marrow transplantation for lymphoma. J Clin Oncol. 1989;7(9):1288-1294. [60] ANURATHAPAN U, PAKAKASAMA S, MEKJARUSKUL P, et al. Outcomes of thalassemia patients undergoing hematopoietic stem cell transplantation by using a standard myeloablative versus a novel reduced-toxicity conditioning regimen according to a new risk stratification. Biol Blood Marrow Transplant. 2014;20(12):2066-2071. [61] SAITO AM, ZAHRIEH D, CUTLER C, et al. Lower costs associated with hematopoietic cell transplantation using reduced intensity vs high-dose regimens for hematological malignancy. Bone Marrow Transplant. 2007;40(3):209-217. |
| [1] | Tao Daiju, Su Haiyu, Wang Yuqi, Shen Zhiqiang, He Bo . Construction and identification of stable PC12 cell lines with high/low expression of miR-122-5p [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1790-1799. |
| [2] | Wu Xianyuan, Zhang Nini, Huang Guilin. Gene transfection technology and tissue fibrosis repair [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(13): 3424-3434. |
| [3] | Guo Daxin, Fan Susu, Zhu Zhendong, Hou Jianhong, Zhang Xuan. Construction of lentiviral vectors for solute carrier family 1 member 5 overexpression and knockdown and stably transfected RAW264.7 cell line [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1414-1421. |
| [4] | Ning Yinkuan, Liu Linzhi, Zhou Cila, Long Yubin. Rabbit bone marrow mesenchymal stem cells transfected with recombinant lentivirus and decalcified bone matrix to construct transgenic tissue engineering materials [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(23): 4851-4858. |
| [5] | Yu Yangyi , Song Zhuoyue, Lian Qiang, Ding Kang, Li Guangheng . AAV-mediated expression of p65shRNA and bone morphogenetic protein 4 synergistically enhances chondrocyte regeneration [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(17): 3537-3547. |
| [6] | Wang Wen, Zheng Pengpeng, Meng Haohao, Liu Hao, Yuan Changyong. Overexpression of Sema3A promotes osteogenic differentiation of dental pulp stem cells and MC3T3-E1 [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(7): 993-999. |
| [7] | Zhang Kefan, Shi Hui. Research status and application prospect of cytokine therapy for osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(6): 961-967. |
| [8] | He Qin, Bu Yan, Lin Guanglei, Luo Jing, Yong Min, Huang Yongqing. Differential expression of RNA binding protein Lin28A regulates osteoblastic differentiation of periodontal ligament stem cells [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(33): 5283-5291. |
| [9] | Huang Yixuan, Du Bin, Liu Xin, Yuan Xinwei, Xi Hongzhong, Guo Mingbin, Mai Jianbin. Research progress and application prospect of polyethyleneimine in bone tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(16): 2563-2570. |
| [10] | Sun Ying, Xiang Guangda, Xu Xiaoli. Effects of myeloid-derived growth factor on ventricular remodeling in aging mice [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(31): 5020-5025. |
| [11] | Li Guangzhao, Chen Rui, Jia Honglin, Ren Liling. Research progress of gene therapy for vascularization of tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(28): 4569-4574. |
| [12] | Gao Zhao, Zhao Yuhao, He Yixiang, Zhao Haiyan, Wang Wenji. DNA hydrogel based on drug delivery and bone tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(27): 4379-4385. |
| [13] | Yuan Lifen, Cao Shuang, Sun Ting. Effect of angiotensinogen-targeted RNA interference in spontaneously hypertensive rats [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(35): 5626-5631. |
| [14] | Wang Weikang, Liu Xiaodong, Zhou Changlin, Tian Jun. Regulatory role of microRNAs in the occurrence and development of osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(35): 5709-5715. |
| [15] | Wei Yunjian, Zhang Fengbo, Long Ping, Jiang Xinxing, Ma Yanlin, Sun Fei, Li Qi. Comparison of different human cell derived induced pluripotent stem cells and embryonic stem cells in tuning embryoid body differentiation [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(25): 4019-4024. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||