Chinese Journal of Tissue Engineering Research ›› 2022, Vol. 26 ›› Issue (14): 2291-2296.doi: 10.12307/2022.497
Pathological changes caused by mutations in polydactyly related genes
Li Qingman, Guo Feng, Liu Fangfang, Zhang Haiying, Liu Hui
- Hainan Key Laboratory for Research and Transformation of Tropical Brain Science, Hainan Medical University, Haikou 571199, Hainan Province, China
-
Received:2021-03-10Revised:2021-03-11Accepted:2021-04-23Online:2022-05-18Published:2021-12-22 -
Contact:Zhang Haiying, MD, Professor, Hainan Key Laboratory for Research and Transformation of Tropical Brain Science, Hainan Medical University, Haikou 571199, Hainan Province, China -
About author:Li Qingman, Master candidate, Hainan Key Laboratory for Research and Transformation of Tropical Brain Science, Hainan Medical University, Haikou 571199, Hainan Province, China -
Supported by:the Natural Science Foundation of Hainan Province, No. 821RC562 (to ZHY); the National Natural Science Foundation of China, No. 81660224 (to ZHY); Innovative Research Project for Graduate Students in Hainan Province, No. Hys2019-288 (to LFF)
CLC Number:
Cite this article
Li Qingman, Guo Feng, Liu Fangfang, Zhang Haiying, Liu Hui. Pathological changes caused by mutations in polydactyly related genes[J]. Chinese Journal of Tissue Engineering Research, 2022, 26(14): 2291-2296.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
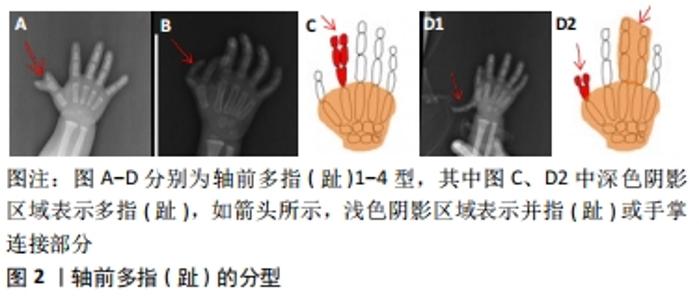
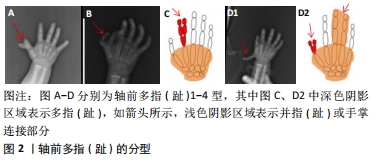
2.1 非综合征型多指(趾)的临床分类 在胚胎发育早期,脊椎动物肢体末端的肢芽沿着3个不同方向的轴发生发展形成指(趾),其特征是细胞有丝分裂活性和程序性细胞死亡之间的动态平衡[4]:包括手指-手腕方向为远近轴、拇指-小指方向为前后轴、手心-手背方向为腹背轴。3个轴向生长所受调控机制不同,中胚层的顶端外胚叶嵴诱导肢芽在远近轴向的生长,极化活动区控制肢芽在前后轴向的发育[5],Wnt信号通路调节肢芽在背腹轴向的延伸[6]。 2.1.1 轴前多指(趾) (1)轴前多指(趾)1型:拇指(趾)多指(趾)表现为拇指(趾)复制1次或2次,形成六指(趾)或七指(趾),见图2A,七指(趾)较为少见,通常为单侧。 (2)轴前多指(趾)2型:拇指(趾)的远节指骨和近节指骨之间有额外的中间指骨,第一掌骨长而薄,两端有骺,见图2B。轴前多指(趾)2型通常是双侧的。 (3) 轴前多指(趾)3型:该类型一般由多个3指(趾)节的示指(趾)代替正常的示指(趾),与示指(趾)相连的掌骨通常发育不全,见图2C。 (4)轴前多指(趾)4型:主要表现为双拇指(趾),末节指(趾)骨呈放射状偏离或拇指(趾)宽且分叉,见图2D1,并指(趾)较为少见[7],见图2D2。"

2.1.2 轴后多指(趾) (1)轴后多指(趾)A型:肢体尺侧或腓骨侧存在发育形态良好的多指(趾),见图3A。 (2) 轴后多指(趾)B型:该型为最常见的多指(趾)畸形,多指(趾)可能发育不良,通常以皮肤赘生物的形式出现,在小拇指(趾)侧形成微小突起或柱状生长,或2.0-3.0 cm长的“蒂”,通常有指甲,见图3B。"

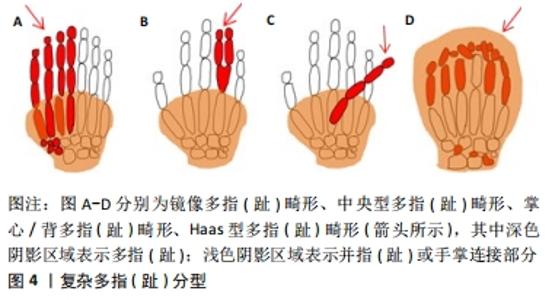
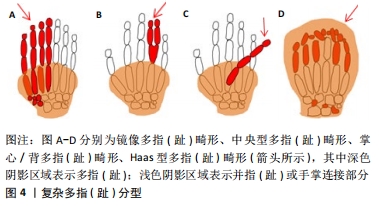
2.1.3 复杂多指(趾) (1)镜像多指(趾)畸形:小指(趾)、无名指(趾)和中指(趾)完全复制并取代拇指(趾)。额外指(趾)的排列是以中心指(趾)开始的降序排列。例如手部:从轴后开始小指、无名指、中指、示指、中指、无名指、小指,见图4A。 (2)中央型多指(趾)畸形:此型表现为无名指(趾)、中指(趾)、示指(趾)的复制,其中无名指(趾)的复制最为常见,示指(趾)次之。畸形常表现为双侧,通常有明显的并指(趾),见图4B。 (3)掌心/背多指(趾)畸形:额外的指(趾)通常出现在掌的腹侧或背侧。它可能表现为发育不良的指(趾)蒂或小皮肤,或发育不良的有(无)指甲的指(趾),见图4C。 (4)Haas型多指(趾)畸形:可表现为轴前或轴后的多指(趾),所有的指(趾)完全“融合”,形成一个“杯子”形状,限制了指(趾)的运动,见图4D。 "
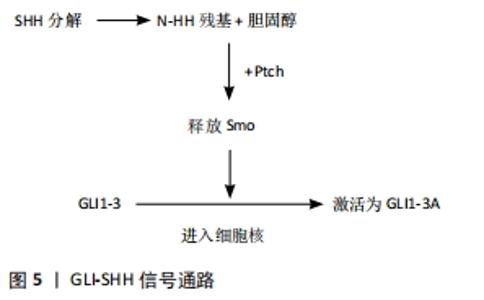
2.2 与多指(趾)相关的基因 GLI3、SHH、TWIST、HOX等基因的突变会导致综合征型和非综合征型的多指(趾)畸形,常见的多指(趾)畸形综合征有Greig综合征、Bardet Biedl综合征、Pallister-Hall综合征和短肋骨多指(趾)综合征等。利用人类基因组序列的完整信息,结合连锁分析、位置克隆、畸形综合征相关染色体异常等多种基因定位的方法,可以鉴定出导致肢体异常的基因位点[4]。 2.2.1 GLI3 GLI3基因位于常染色体7p14.1,由14个外显子组成,其中包括一个大于2 500 bp的大外显子。GLI3基因编码锌指转录因子,在hedgehog(Hh)信号转导通路中发挥作用,可以影响肢体发育。RADHAKRISHNA等[8]发现GLI3基因突变会引起轴后多指(趾)A1型,在该型患者中发现GLI3等位基因的第764位密码子发生突变,突变发生在锌指结构的保守序列中。FURNISS等[9]在轴后多指(趾)B型患者中,发现GLI3基因中缺失1-bp的杂合移码突变。AL-QATTAN等[10]在轴后多指(趾)畸形患者中发现,GLI3基因中出现2bp缺失的杂合移码突变。 GLI3基因外显子缺失、错义突变以及中间段的截断突变导致了Pallister-Hall综合征[11], Pallister-Hall综合征为常染色体显性遗传,典型特征为并多指(趾)和下丘脑错构瘤,其他特征包括会厌裂、肛门闭锁、生长激素缺乏、垂体功能减退和生殖器发育不全。Pallister-Hall综合征患者中GLI3基因发生移码突变,线粒体DNA的许多片段在进化过程中会从线粒体基因组迁移到核基因组[12],这导致线粒体蛋白质组中的绝大多数蛋白质由核基因组编码,线粒体DNA移位会导致GLI3基因功能中断,从而引起特殊的临床特征。而中间段以外的截断突变导致了Greig头足综合征,Greig头足综合征为常染色体显性遗传,典型特征是头大、眼间距加宽、轴前多趾,部分可见轴后多趾和皮肤并趾,发育迟缓、智力障碍或癫痫比较少见(<10%),大约20%的Greig头足综合征患者有胼胝体发育不全或不全[13]。Greig头足综合征患者中GLI3基因发生大量缺失或易位[14],对Greig头足综合征患者的GLI3基因进行突变筛查,发现15个新的突变存在于GLI3基因中[15],这些突变分布在编码基因的各个区域,其中大多数是截短突变(15个突变中的9个)。新的突变导致蛋白质转录过程提前终止,主要发生在编码DNA结合域中心区域的N端或中间部分[16]。 2.2.2 SHH SHH基因定位于常染色体7q36.3,是Hh家族中的一员,仅在四肢间充质后缘的极化活动区内表达,受肢体极化区增强调控序列的调控元件驱动[17]。Hh信号通路高度保守,在胚胎骨发育和出生后的骨重建中起着关键作用,该信号失调可导致肢体畸形,如多指(趾)畸形[18]。SHH正常在轴后表达,而轴前异位表达与轴前多指(趾)畸形有关。极化区增强调控序列是SHH顺式调控元件,位于SHH基因上游1 Mb处,能促进肢体的发育,极化区增强调控序列功能增强或拷贝数增加会导致非综合征性轴前多指(趾)[19-20];成纤维细胞生长因子10缺失时,无法形成顶端外胚叶嵴和极化活动区,可能会导致四肢部分完全缺失[4]。SHH编辑的蛋白在亨氏节、神经管的底板、早期肠内胚层、肢芽的后部和整个脊索中均表达[21],因此,SHH可能在中枢神经系统和肢体发育过程中起重要作用。 SHH、FGF4和骨形态发生蛋白拮抗剂(gremlin)形成SHH-FGF4正反馈通路,可促进脊椎动物肢体远端发育。在极化活动区中生成的SHH信号可调节FGF4,FGF4反过来可维持SHH-FGF4反馈通路。肢体正常发育时,gremlin将SHH信号从极化活动区传递到顶端外胚叶嵴[22],使其与FGF4形成正常反馈回路。反之,当gremlin表达量减少,不能将SHH信号从极化活动区传递到顶端外胚叶嵴,无法正常建立SHH-FGF4反馈环,会造成多指(趾)畸形。研究发现将表达gremlin的细胞移植到突变肢芽中则恢复了FGF4的表达,并重新建立了SHH-FGF4反馈通路,该研究揭示了gremlin激活后,可以诱导FGF4表达并建立SHH-FGF4反馈回路[23]。与SHH基因相关的几个转录因子,如Hand2、GLI3、gremlin和骨形态发生蛋白等的改变会导致多指(趾)畸形[24]。 SHH基因突变所引起的多指(趾)综合征型包括足三趾拇趾-多趾综合征[25]、Bardet-Biedl综合征[26]、胸大肌综合征、Laurence Moon Bardet Biedl综合征等[27]。与Hh信号通路相关的综合征有很多,包括大头畸形、肿瘤、肢体和面部畸形等特征。而几乎每一种Hh信号通路相关的遗传综合征都有肢体畸形表型,其中多指(趾)最为常见[28]。 Bardet-Biedl综合征是一种异质性常染色体隐性遗传疾病,其临床特征表现为肥胖、视网膜病、多指(趾)畸形、肾脏畸形、学习障碍和生殖功能低下,同时还可能存在糖尿病和高血压等的继发性表型。迄今为止,已有16个BBS基因被证明能够独立引起这种疾病。7种BBS蛋白(BBS1、BBS2、BBS4、BBS5、BBS7、BBS8和BBS9)可以与相互作用蛋白BBIP10结合形成BBSome复合物,它可以帮助Smo和Ptch1转运到细胞内,通过SHH-GLI3信号通路促进下游靶基因转录。BBS基因缺失时Smo和Ptch1在纤毛中累积,使SHH-GLI3通路受阻,从而导致多指(趾)畸形[29]。 2.2.3 TWI TWI基因位于常染色体7p21.1,在中胚层发育中起着至关重要的作用,尤其在肢体和颅面部发育中,TWIST1在发育过程中转录调控的分子机制尚不清楚。TWIST1基因编码一个基本的螺旋-环-螺旋转录因子,和其他转录因子形成二聚体或异二聚体调节靶基因的表达[30],如TWIST1和心脏神经脊衍生物表达转录因子2(Hand2)形成的二聚体决定了正常的骨形态发生,其表达比例的不平衡与多指(趾)畸形的发病机制有关[31]。HIRSCH等[32]研究发现,TWIST1基因中的增强子突变也可能导致TWIST1表达的减少,从而产生与TWIST1基因突变相同的表型结果。 TWIST1基因突变包括错义、无义、小片段的插入和删除与大片段的插入和删除。在这些突变中,大多数无义突变(84.0%)和小片段的插入和删除(95.7%),通常发生在TA结构域的上游,导致提前产生了终止密码子[33]。突变会导致Saethre-Chotzen综合征,临床表现为面部不对称、斜视、上睑下垂和耳朵的特征性外观(具有突出的上和/或下小腿的小耳廓),可出现并指(趾)或少指(趾) [34]。个别Saethre-Chotzen综合征患者表现的非典型症状为智力障碍、发育迟缓和学习困难。 在体外研究中,PKA和PP2A-B56δ可以调节TWIST1和Hand2b二聚体残基磷酸化,这些残基的磷酸化和磷酸化模拟物可以改变二聚体亲和力,导致磷酸调节环路失调[33];在Saethre-Chotzen综合征患者中发现TWIST1基因突变,该突变可导致磷酸环路调节失调。研究表明,TWIST1和Hand2表达比例对肢体的正常发育至关重要,它们相互作用需要通过磷酸化的螺旋碱基Ⅰ调节,TWIST1表达降低或Hand2表达增加都会导致多指(趾)畸形[35]。 2.2.4 HOX 人类染色体中有39个HOX基因,其中HOXA13和HOXD13与多指(趾)和并指(趾)有密切联系。HOX基因编码一组高度保守的转录因子,它们控制着细胞在身体特定区域的发生发展。研究转基因小鼠的实验发现,HOXA和HOXD基因5′端的靶向突变和过度表达不仅会改变实验小鼠四肢的大小、形状和数量,而且会延迟软骨化和骨化[36]。上文所述的SHH基因和HOX基因之间的相互转录调控以及HOXD12与GLI3R的结合,均会导致GLI3R活性的抑制[33]。当去除HOXA13和HOXD11-13的一个功能等位基因时,手指的个数从7-9个增加到8-9个;当HOXD11-13的2个等位基因都被移除时,手指个数增加到了9-11个[37]。 (1)HOXA13:HOXA13基因定位于常染色体7p15.2,研究发现该蛋白的表达与指(趾)远端位点有关,它是激活远端特异基因表达所必需的。染色质构象分析显示,5个HOXA基因(HOXA9-13)位于染色质亚拓扑结构域内,在远端肢体中表达,与结构域中的增强子相互作用。HOXA11在指(趾)近端表达受限,与基因本身的特征有关,并且HOXA11是调节远端肢体的一个重要因素。HOXA11as-b中的增强子,可以控制HOXA11as-b的表达,增强子的缺失会导致HOXA11as-b在远端不表达[38]。 当远端HOX基因被移除时,手指数量增加,但手掌大小不变,这是因为手指变细,彼此之间间隙更窄,同时保持规则间隔[37],因此在手掌大小不变的情况下容纳更多手指。这与GLI3基因单突变体机制相似。 (2)HOXD13:HOXD13基因定位于常染色体2q31.1。HOXD10-13基因缺失导致细胞中的HOXD9表达量增加,此时SHH表达减少,这表明HOXD10-13的表达对诱导SHH表达至关重要。在没有HOXD13的情况下,HOXD11在特定区域表达增加会导致多指(趾)畸形,而HOXD10和HOXD12的增加对指(趾)却没有影响。 研究表明,HOXD13氨基末端区域插入短聚丙氨酸片段导致并(多)指(趾)(synpolydactyly,SPD)。SPD有3种类型:SPD1临床表现为中央并(多)指(趾),轴后并(多)指(趾);SPD2临床表现为中央及轴后并(多)指(趾),轴后并指(趾);SPD3临床表现为中央并(多)指(趾),轴后并(多)指(趾)[39]。SPD2和SPD3这两种类型极为少见,这里主要介绍SPD1。在SPD1中,通过全基因组测序来鉴定HOXD13的突变位点,结果表明HOXD13外显子1中有24个碱基对拷贝数目增加,根据这些突变预测在HOXD13蛋白的N末端结构域中有8个额外的丙氨酸残基[40]。研究发现HOXD13基因中的一个错义突变,会产生一种独特的短指(趾)-多指(趾)综合征,除了典型和非典型性SPD特征外还包括手中央多指畸形,足部第三趾骨发育不全,也有其他特殊症状,如指(趾)甲和远端指骨发育不全或缺失,第三至第五掌骨和跖骨短、尺骨茎突发育不全等[36]。 2.3 多指(趾)的发生机制 2.3.1 GLI3-SHH信号通路 GLI3-SHH是一种进化保守的通路,在胚胎骨发育和出生后的骨重建中起着关键作用[41]。在哺乳动物细胞中,Ptch1和Smoothened(Smo)是GLI3-SHH通路的另外2个调控因子,Ptch1位于纤毛上,对Smo有抑制作用。GLI3蛋白是调控Hh信号通路的关键分子,是一种双功能转录因子,降解后产生2种不同作用的蛋白,C末端为GLI3A转录激活物,N末端为GLI3R阻遏物。无SHH时,Smo被Ptch1抑制,GLI1-3不能进入细胞核,胞质中的GLI2和GLI3通过SUFU(融合抑制因子)复合体结合,并被蛋白激酶A磷酸化,导致它们裂解为GLIR和GLIA,随后进入细胞核,抑制下游靶基因的转录,阻断了GLI3-SHH信号通路[42-43]。有SHH时,SHH与Ptch1结合,使Ptch1对Smo的抑制作用消失,GLI1-3进入细胞核,使GLI3R被激活成GLI3A形式,促进下游靶基因的转录[44],见图5。这两种形式的GLI3R和GLI3A以及它们在GLI3R/GLI3A中所占的比例直接与指(趾)的数量和形态有关[45]。"

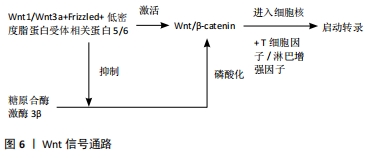
2.3.2 Wnt信号通路 Wnt信号在细胞增殖、细胞命运决定和分化的各种发育过程中都有涉及[46]。Wnt与受体复合物结合并激活2个信号通路,即β-catenin依赖的典型通路和β-catenin不依赖的非典型通路。β-catenin依赖的Wnt通路(Wnt/β-catenin)能够调节软骨细胞分化、生长板组装和软骨完整性[47];Wnt缺失时,糖原合酶激酶3β使细胞中的β-catenin磷酸化,并通过泛素-蛋白酶体途径降解[48];Wnt存在时,Wnt与受体复合物抑制糖原合酶激酶3β的活性并激活Wnt/β-catenin通路,进而诱导β-catenin在细胞质中的积累[49-50],最终导致其转位到细胞核,在胞核与T细胞因子/淋巴样增强因子相互作用,启动靶基因的转录,见图6。"

低密度脂蛋白受体相关蛋白5/6复合物是Wnt/β-catenin通路的重要调节因子,它与硬化蛋白(Wnt拮抗剂,由骨细胞分泌)结合,可以抑制Wnt/β-catenin信号转导[51]。纤毛内转运系统由纤毛内转运蛋白复合物A,B组成,可沿纤毛轴做双向运动,纤毛内转运蛋白80、纤毛内转运蛋白88蛋白是纤毛内转运蛋白复合物B的核心蛋白[50-53],纤毛内转运蛋白140蛋白是纤毛内转运蛋白复合物A的核心蛋白[54],纤毛内转运蛋白基因突变导致纤毛病,表现为多种骨骼异常[55-57],例如纤毛内转运蛋白80蛋白部分突变会引起人类Jeune窒息性胸营养不良和短肋骨多指(趾)综合征Ⅲ型、Ⅳ型[58]。 2.3.3 信号通路之间会相互影响 研究发现,当Ptch1缺失时,Hh信号通路可能通过GLI3激活Wnt5a/6的表达,导致β-catenin活性增强[59-61],Hh信号通路与Wnt信号通路相互影响,推测Wnt和Hh信号通路之间可能存在串扰。敲降纤毛内转运蛋白80基因可下调Hh信号活性,而上调Wnt信号活性,在纤毛内转运蛋白80基因沉默的细胞中过表达GLI2基因促进了软骨形成,并恢复了纤毛内转运蛋白80沉默后的软骨缺陷[52],这些发现提示了纤毛内转运蛋白80调控软骨细胞分化过程与Hh和Wnt信号通路之间存在某种紧密联系,因此,研究某一个基因突变对认识多指(趾)畸形是有局限性的。近年来,蛋白组学与基因病的结合越来越密切,它可以大规模地研究蛋白质的特征,包括蛋白质的表达水平,转录、翻译后的修饰,蛋白与蛋白相互作用等;可以通过蛋白组学发现更多与多指(趾)畸形相关的信号通路,从通路入手,研究通路中的相关蛋白,并进一步研究通路之间的相互影响;对基因的位置关系、作用位点、生物功能等方面进行更深入的探讨,以期更加全面的了解多指(趾)畸形的发生机制。"

| [1] 赵献芝, 白小青, 左福元. 多指(趾)基因研究进展[J]. 上海畜牧兽医通讯,2007,52(1):5-6. [2] VERMA PK, EL-HAROUNI AA. Review of literature: genes related to postaxial polydactyly. Front Pediatr. 2015;3:8. [3] ZAIB T, JI W, SALEEM K, et al. A heterozygous duplication variant of the HOXD13 gene caused synpolydactyly type 1 with variable expressivity in a Chinese family. BMC Med Genet. 2019;20(1):203. [4] PHADKE SR, SANKAR VH. Polydactyly and genes. Indian J Pediatr. 2010; 77(3):277-281. [5] LETTICE LA, WILLIAMSON I, WILTSHIRE JH, et al. Opposing functions of the ETS factor family Ddefine SHH spatial expression in limb buds and underlie polydactyly. Development Cell. 2012;22(2):459-467. [6] WANG T, XUAN ZP, DOU YC, et al. Identification of novel mutations in preaxial polydactyly patients through whole-exome sequencing.Mol Genet Genomic Med. 2019;7(6):e690. [7] BURGER EB, BAAS M, HOVIUS SER. A Jeannette M Hoogeboom, Christianne A van Nieuwenhoven. Preaxial polydactyly of the foot. Acta Orthop. 2018;89(1):113-118. [8] RADHAKRISHNA U, BLOUIN JL, MEHENNI H, et al. Mapping one form of autosomal dominant postaxial polydactyly type A to chromosome 7p15-q11.23 by linkage analysis. Am J Hum Genet. 1997;60:597-604. [9] FURNISS D, CRITCHLEY P, GIELE H, et al. Nonsense-mediated decay and the molecular pathogenesis of mutations in SALL1 and GLI3. Am J Med Genet A. 2007;143(24):3150-3160. [10] AL-QATTAN MM. A novel frameshift mutation of the GLI3 gene in a family with broad thumbs with/without big toes, postaxial polydactyly and variable syndactyly of the hands/feet. Clin Genet. 2012;82(5):502-504. [11] CHANDRA SR, DARYAPPA MM , MUDABBIR MAM, et al. Pallister-Hall Syndrome. J Pediatr Neurosci. 2017;12(3):276-279. [12] WOISCHNIK M, MORAES CT. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 2002;12:885-893. [13] BIESECKER LG, JOHNSTON JJ, ADAM MP, et al. Greig Cephalopolysyndactyly Syndrome. University of Washington. 2020. [14] PETTIGRE AL, GREENBERG F, CASKEY CT, et al. Greig syndrome associated with an interstitial deletion of 7p: confirmation of the localization of Greig syndrome to 7p13. Hum Genet. 1991;87:452-456. [15] SUSKE MK, WILD A, TOPP J, et al. Point mutations throughout the GLI3 gene cause Greig cephalopolysyndactyly syndrome. Hum Molec Genet. 1999;8:1769-1777. [16] UNNI JV, DARYANI D, SREEJAN KC, et al. Greig Cephalopolysyndactyly Syndrome with Oral Manifestations: A Rare Case Report. Int J Appl Basic Med Res. 2020;10(2):140-142. [17] WANG B, DIAO YT, LIU QJ, et al. An increased duplication of ZRS region that caused more than one supernumerary digits preaxial polydactyly in a large Chinese family. Sci Rep. 2016;6:38500. [18] ITO S, KITAZAWA R, HARAGUCHI R, et al. Novel GLI3 variant causing overlapped Greig cephalopolysyndactyly syndrome (GCPS) and Pallister-Hall syndrome (PHS) phenotype with agenesis of gallbladder and pancreas. Diagn Pathol. 2018;13(1):1. [19] AL-QATTAN MM. Zone of polarizing activity regulatory sequence mutations/duplications with preaxial polydactyly and longitudinal preaxial ray deficiency in the phenotype: a review of human cases, animal models, and insights regarding the pathogenesis. Biomed Res Int. 2018;2018:1573871. [20] PETIT F, JOURDAIN AS, HOLDER-ESPINASSE M, et al. The disruption of a novel limb cis-regulatory element of SHH is associated with autosomal dominant preaxial polydactyly hypertrichosis. Eur J Hum Genet. 2016; 24(1):37-43. [21] ROELINK H, AUGSBURGER A, HEEMSKERK J, et al. Floorplate and moyor neuron induction by VHH-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76(4):761-775. [22] NACU E, GROMBERG E, OLIVEIRA CR, et al. FGF8 and SHH substitute for anterior-posterior tissue interactions to induce limb regeneration. Nature. 2016;533(7603):407-410. [23] Zúñiga A, Haramis AP, McMahon AP, et al. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature. 1999;401(6753):598-602. [24] JACQUELINE L, NORRIEA, LEWANDOWSKI JP, et al. Dynamics of BMP signaling in limb bud mesenchyme and polydactyly. Dev Biol. 2014; 393(2):270-281. [25] LIU ZH, YIN N, GONG LH, et al. Microduplication of 7q36.3 encompassing the SHH long-range regulator(ZRS) in a patient with triphalangeal thumb-polysyndactyly syndrome and congenital heart disease. Mol Med Rep. 2017;15:793-797. [26] HULLEMAN JD, NGUYEN A, RAMPRASAD VL, et al. A novel H395R mutation in MKKS/BBS6 causes retinitis pigmentosa and polydactyly without other findings of Bardet-Biedl or McKusick-Kaufman ayndrome. Mol Vis. 2016;22:73-81. [27] OMAIR A, KHAN, MAJEED R, et al. Rarity of laurence moon bardet biedl syndrome and its poor management in the pakistani population. Cureus. 2019;11(2):e4114. [28] KLEIN S D, NGUYEN D C, BHAKTA V, et al. Mutations in the sonic hedgehog pathway cause macrocephaly-associated conditions due to crosstalk to the PI3K/AKT/mTOR pathway. Am J Med Genet A. 2019;179(12):2517-2531. [29] ZHANG QH, SEO S, BUGGE K, et al. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet. 2012;21(9):1945-1953. [30] LOEBEL DA, HOR AC, BILDSOE H, et al. Regionalized TWIST1 activity in the forelimb bud drives the morphogenesis of theproximal and preaxial skeleton. Dev Biol. 2012;362(2):132-140. [31] Firulli BA, KRAWCHUK D, CENTONZE VE, et al. Altered TWIST1 and HAND2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37(4):373-381. [32] HIRSCH N, ESHEL R, YAACOV RB, et al. Unraveling the transcriptional regulation of TWIST1 in limb development. PLoS Genet. 2018;14(10): e1007738. [33] CHEN RZ, CHENG XB, TAN YX, et al. An ENU-induced mutation in TWIST1 transactivation domain causes hindlimb polydactyly with complete penetrance and dominant-negatively impairs E2A-dependent transcription. Sci Rep. 2020;10(1):2501. [34] PELC A, MIKULEWICZ M. Saethre-Chotzen Syndrome: Case report and literature review. Dent Med Probl. 2018;55(2):217-225. [35] ZHANG Z, SUI PF, DONG AW, et al. Preaxial polydactyly: interactions among ETV, TWIST1 and HAND2 control anterior-posterior patterning of the limb. Development. 2010;137(20):3417-3426. [36] CARONIA G, GOODMAN FR, MCKEOWN CM, et al. An I47L substitution in the HOXD13 homeodomain causes a novel human limb malformation by producing a selective loss of function. Development. 2003;130(8):1701-1712. [37] SHETH R, MARCON L, BASTIDA MF, et al. HOX genes regulate digit patterning by controlling the wavelength of a Turing-type mechanism. Science. 2012; 338(6113):1476-1480. [38] KHERDJEMIL Y, LALONDE RL, SHETH R, et al. Evolution of HOXA11 regulation in vertebrates is linked to the pentadactyl state. Nature. 2016;539(7627): 89-92. [39] MALIK S, GRZESCHIK KH. Synpolydactyly: clinical and molecular advance. Clin Genet. 2008;73(2):113-120. [40] ROGERS BH, SCHMIEG SL, PEHNKE ME, et al. Evaluation and management of preaxial polydactyly. Curr Rev Musculoskelet Med. 2020;13(4):545-551. [41] LEX RK, JI ZC, FALKENSTEIN KN, et al. GLI transcriptional repression regulates tissue-specific enhancer activity in response to Hedgehog signaling. Elife. 2020;9:e50670. [42] RUBINO S, QIAN J, PINHEIRO-NETO CD, et al. A familial syndrome of hypothalamic hamartomas, polydactyly, and SMO mutations: a clinical report of 2 cases. J Neurosurg Pediatr. 2018;23(1)98-103. [43] HARAGUCHI R, KITAZAWA R, KOHARA Y. Recent Insights into Long Bone Development: Central Role of Hedgehog Signaling Pathway in Regulating Growth Plate. INT J Mol Sci. 2019;20(23):5840. [44] CHEN W, REN XR, NELSON CD, et al. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science. 2004; 306:2257-2260. [45] Verma PK, EL-HAROUNI AA. Review of literature: genes related to postaxial polydactyly. Front Pediatr. 2015;3:8. [46] PARRA-TORRES AY, ENRÍQUEZ J, JIMÉNEZ-ORTEGA RF. Expression profiles of the Wnt/β catenin signaling related extracellular antagonists during proliferation and differentiation in human osteoblast like cells. Exp Ther Med. 2020;20(6):254. [47] TAMAMURA Y, OTANI T, KANATAIN N, et al. Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. 2005;280: 19185-19195. [48] LOGAN CY, NUSSE R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781-810. [49] GORDON MD, NUSSE R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281: 22429-22433. [50] MOSIMANN C, HAUSMANN G, BASLER K. β-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol. Cell Biol. 2009; 10:276-286. [51] LI X, ZHANG Y , KANG H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883-19887. [52] LI XH, YANG ST, HAN L, et al. Ciliary IFT80 is essential for intervertebral disc development and maintenance. FASEB. 2020;34(5):6741-6756. [53] LIU M, ALHARBI M, GRAVES D, et al. IFT80 is required for fracture healing through controlling the regulation of TGFβ signaling in chondrocyte differentiation and function. J Bone Miner Res. 2020;35(3):571-582. [54] ZHANG CY, ZHANG S, SUN Y. Expression of IFT140 During Bone Development. J Histochem. 2019;67(10):723-734. [55] LIM J, LI X H, YUAN X, et al. Primary cilia control cell alignment and patterning in bone development via ceramide-PKCζ-β-catenin signaling. Commun Biol. 2020;3(1):45. [56] HE X. Cilia put a brake on Wnt signalling. Nat Cell Biol. 2008;10:11-13. [57] CHANG CF, SERRA R. Ift88 regulates hedgehog signaling, Sfrp5 expression, and beta-catenin activity in post-natal growth plate. J Orthop Res. 2013; 31(3):350-356. [58] BIZAOUI V, HUBER C, KOHAUT E, et al. Mutations in IFT80 cause SRPS Type IV. Report of two families and review. Am J Med Genet A. 2019;179(4):639-644. [59] DENG Q, LI P, CHE MJ, et al. Activation of hedgehog signaling in mesenchymal stem cells induces cartilageand bone tumor formation via Wnt/b-Catenin. Elife. 2019;8:e50208. [60] WANG Y, ZENG HQ, LIU AM. Distinct Activities of Gli1 and Gli2 in the Absence of Ift88 and the Primary Cilia. J Dev Biol. 2019;7(1):5. [61] EGGENSCHWILER JT, ANDERSON KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345-373. |
| [1] | Bao Xianguo, Gao Zengxin, Wu Zhanpo, Chen Youmin, Cheng Qinghua, Lu Haitao, Guo Changzheng, Xu Shuai. Correlation between lumbar posterior muscle and local kyphosis in patients with degenerative thoracolumbar kyphosis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1418-1423. |
| [2] | Xue Yadong, Zhou Xinshe, Pei Lijia, Meng Fanyu, Li Jian, Wang Jinzi . Reconstruction of Paprosky III type acetabular defect by autogenous iliac bone block combined with titanium plate: providing a strong initial fixation for the prosthesis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1424-1428. |
| [3] | Zhu Chan, Han Xuke, Yao Chengjiao, Zhou Qian, Zhang Qiang, Chen Qiu. Human salivary components and osteoporosis/osteopenia [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1439-1444. |
| [4] | Zhang Haobo, Zhao Yunan, Yang Xuejun. Role and therapeutic implications of pyroptosis in intervertebral disc degeneration [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1445-1451. |
| [5] | Jin Tao, Liu Lin, Zhu Xiaoyan, Shi Yucong, Niu Jianxiong, Zhang Tongtong, Wu Shujin, Yang Qingshan. Osteoarthritis and mitochondrial abnormalities [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1452-1458. |
| [6] | Zhang Lichuang, Xu Hao, Ma Yinghui, Xiong Mengting, Han Haihui, Bao Jiamin, Zhai Weitao, Liang Qianqian. Mechanism and prospects of regulating lymphatic reflux function in the treatment of rheumatoid arthritis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1459-1466. |
| [7] | Wang Baojuan, Zheng Shuguang, Zhang Qi, Li Tianyang. Miao medicine fumigation can delay extracellular matrix destruction in a rabbit model of knee osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1180-1186. |
| [8] | Li Zhiyi, He Pengcheng, Bian Tianyue, Xiao Yuxia, Gao Lu, Liu Huasheng. Bibliometric and visualized analysis of ferroptosis mechanism research [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1202-1209. |
| [9] | Wang Jing, Xiong Shan, Cao Jin, Feng Linwei, Wang Xin. Role and mechanism of interleukin-3 in bone metabolism [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1260-1265. |
| [10] | Zhu Chan, Han Xuke, Yao Chengjiao, Zhang Qiang, Liu Jing, Shao Ming. Acupuncture for Parkinson’s disease: an insight into the action mechanism in animal experiments [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(8): 1272-1277. |
| [11] | Tian Chuan, Zhu Xiangqing, Yang Zailing, Yan Donghai, Li Ye, Wang Yanying, Yang Yukun, He Jie, Lü Guanke, Cai Xuemin, Shu Liping, He Zhixu, Pan Xinghua. Bone marrow mesenchymal stem cells regulate ovarian aging in macaques [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 985-991. |
| [12] | Guo Jia, Ding Qionghua, Liu Ze, Lü Siyi, Zhou Quancheng, Gao Yuhua, Bai Chunyu. Biological characteristics and immunoregulation of exosomes derived from mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1093-1101. |
| [13] | Wu Weiyue, Guo Xiaodong, Bao Chongyun. Application of engineered exosomes in bone repair and regeneration [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1102-1106. |
| [14] | Zhou Hongqin, Wu Dandan, Yang Kun, Liu Qi. Exosomes that deliver specific miRNAs can regulate osteogenesis and promote angiogenesis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1107-1112. |
| [15] | Zhang Jinglin, Leng Min, Zhu Boheng, Wang Hong. Mechanism and application of stem cell-derived exosomes in promoting diabetic wound healing [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1113-1118. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||