Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (24): 6306-6315.doi: 10.12307/2026.367
Previous Articles Next Articles
Animal models of neurogenic heterotopic ossification: key disease progression and pathogenesis
Ren Qingsong1, Xie Yulei1, 2, Liu Jingjing1, Lin Jingyi1, Long Danlei1, Zhang Chunyu3, Xie Liang1, Zheng Kaiyuan1, Wang Yinxu1
- 1Department of Rehabilitation Medicine, Affiliated Hospital of North Sichuan Medical College, Nanchong 637000, Sichuan Province, China; 2School of Sports Medicine and Rehabilitation, Affiliated Hospital of North Sichuan Medical College, Nanchong 637000, Sichuan Province, China; 3Department of Cardiology, Affiliated Hospital of Southwest Medical University, Luzhou 646000, Sichuan Province, China
-
Received:2025-06-06Revised:2025-09-15Online:2026-08-28Published:2026-02-03 -
Contact:Wang Yinxu, PhD, Associate chief physician, Department of Rehabilitation Medicine, Affiliated Hospital of North Sichuan Medical College, Nanchong 637000, Sichuan Province, China -
About author:Ren Qingsong, MS candidate, Department of Rehabilitation Medicine, Affiliated Hospital of North Sichuan Medical College, Nanchong 637000, Sichuan Province, China -
Supported by:Sichuan Province Science and Technology Innovation Seed Project, No. MZGC20230044 (to ZKY); North Sichuan Medical College Youth Project - Natural Science Category, No. 2023JC011, CBY22-QNA45 (to XYL)
CLC Number:
Cite this article
Ren Qingsong, Xie Yulei, Liu Jingjing, Lin Jingyi, Long Danlei, Zhang Chunyu, Xie Liang, Zheng Kaiyuan, Wang Yinxu. Animal models of neurogenic heterotopic ossification: key disease progression and pathogenesis[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(24): 6306-6315.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
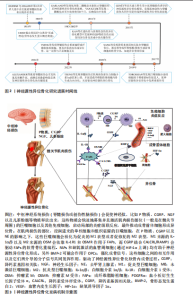
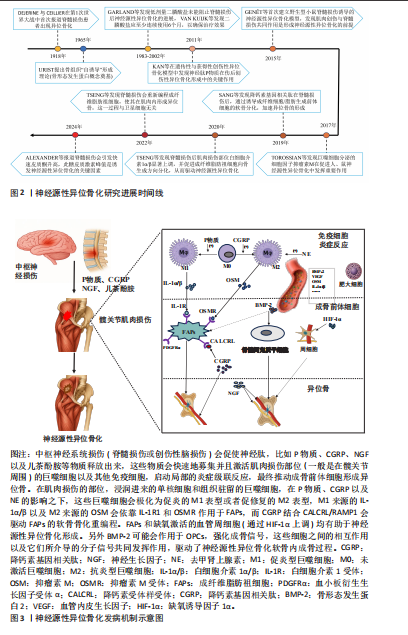
神经源性异位骨化是中枢神经系统损伤后常见的并发症,其研究过程随着临床观察的深入以及基础机制的探索,逐渐取得了显著进展。图2系统整理了神经源性异位骨化研究领域的关键里程碑,呈现出神经源性异位骨化从临床发现直至分子机制阐释的发展进程,自1918年DEJERINE与CéILLIER首次报告脊髓损伤后出现异位骨化以来,神经源性异位骨化渐渐受到临床关注。1965年,URIST提出了与骨形成相关的“骨形态蛋白”,也就是骨形态发生蛋白的概念,为分子机制研究奠定了基础;2011年,VAN KUIJK等开展了低剂量二膦酸盐预防神经源性异位骨化的临床试验,为干预手段提供了一定支持。近些年,多个动物模型研究对发病机制以及干预策略展开了探索,时间轴完整展示了神经源性异位骨化从临床观察到动物模型建立再到分子机制阐释的演进过程。 要深入剖析神经源性异位骨化的发病机制,需从其复杂的细胞和分子相互作用网络着手,图3呈现了中枢神经系统损伤引发局部组织异位骨形成的细胞及分子机制,左侧部分说明神经损伤会开启炎症级联反应,随后募集并激活免疫细胞,最终促使成骨前体细胞形成异位骨,右侧部分则详细阐述了损伤部位的巨噬细胞、成纤维脂肪祖细胞等细胞在神经肽、细胞因子等因素影响下的极化与重编程过程,这些细胞依靠协同合作实现软骨内成骨。 2.1 神经源性异位骨化的流行病学、临床表现与临床干预挑战 据统计,全球约有2 000万脊髓损伤患者,其中神经源性异位骨化的发生率可达10%-40%,且多在损伤后1-3个月内出现;相比之下,在创伤性脑损伤患者中,神经源性异位骨化的发生率为10%-23%[2,7-9]。神经源性异位骨化的解剖分布具有显著偏好性,约60.9%发生于髋关节周围肌群,其次为肘关节(21.3%)和膝关节(14.3%)[3],其发病风险与损伤严重程度呈正相关:完全性脊髓损伤患者发生神经源性异位骨化的风险是不完全损伤者的2-4倍[10-12],且颈段和胸段损伤患者比腰段患者更容易发生神经源性异位骨化,可能与交感神经系统的损伤程度有关[13]。此外,20-30岁的年轻男性更易发生神经源性异位骨化,推测与性激素介导的炎症反应增强有关[14-17];同时,携带ACVR1基因R206H突变的个体对骨形态发生蛋白信号通路更为敏感,其发生神经源性异位骨化的风险为普通人群的3.2倍[18]。康复训练启动延迟会加剧局部炎症和肌肉萎缩,进一步增加异位骨形成风险[9];若脊髓减压术延迟超过24 h实施,神经源性异位骨化风险增加3.4倍[19]。在脊髓损伤患者中,痉挛(OR=3.8)、压疮(OR=2.1)、损伤后时间(OR=1.1)和深静脉血栓均显著增加神经源性异位骨化风险[20-21];而在创伤性脑损伤患者中,较低的功能性步行能力量表评分、神经源性膀胱和全身感染也是独立的危险因素[22]。 神经源性异位骨化的局部表现主要包括关节活动度受限、疼痛及血管神经受压。影像学显示,异位骨组织可包裹神经血管束,导致下肢痉挛加重及深静脉血栓风险升高1.8-2.0倍[17]。长期的活动范围受限进一步引发肌肉萎缩和关节僵硬,其中髋关节神经源性异位骨化患者的最大关节活动范围较损伤初期下降超过50%[23-25]。在全身层面,神经源性异位骨化常伴随长时间的炎症高反应状态(如白细胞介素6、C-反应蛋白增高)及内分泌紊乱(皮质醇节律紊乱、褪黑素显著减少81%),形成“神经损伤-炎症-骨代谢失衡”的恶性循环[26-27]。 当前临床干预面临双重困境:预防层面,"
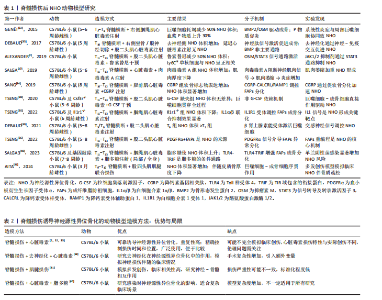
非类固醇抗炎药(如吲哚美辛)虽可降低神经源性异位骨化的发生率,但存在胃肠道损伤及骨折愈合延迟等不良反应[2-3,28];治疗层面,超过1/3患者对药物无显著应答,常需依赖手术切除,但术后复发率高达19.8%-50%,且通常要等待骨化灶成熟(18-24个月)才可手术,进一步延误康复并加重经济负担[3,9,29-30]。此外,神经源性异位骨化与深静脉血栓的相互作用可形成“深静脉血栓-神经源性异位骨化”恶性循环,进一步增加治疗难度[8,31]。 神经源性异位骨化的病理特征与临床危害凸显了它作为中枢神经系统损伤后严重并发症的重要性,亟需通过早期预警和靶向干预改善患者预后。 2.2 神经源性异位骨化的动物模型研究进展 神经源性异位骨化是一种中枢神经损伤后的复杂病理现象,涉及中枢与外周病理的多层次交互。尽管神经源性异位骨化的发病机制尚未完全明确,但目前已开发了多种动物模型,成功重现了神经源性异位骨化的部分关键病理特征,为探索其分子与细胞机制以及开发治疗策略提供了重要依据。其中,脊髓损伤诱导神经源性异位骨化模型强调了肌肉炎症、去神经化及局部炎症放大在疾病发生中的作用;另一方面,创伤性脑损伤诱导的神经源性异位骨化模型则突出了系统性神经信号(如降钙素基因相关肽和肾上腺素)对成骨微环境的调控。 2.2.1 脊髓损伤后神经源性异位骨化模型 脊髓损伤后神经源性异位骨化模型主要通过模拟脊髓损伤后局部肌肉炎症与系统性神经信号失调的联合作用,成功重现了神经源性异位骨化的病理特征,并揭示了神经、免疫及成骨微环境之间的复杂交互作用[32]。脊髓损伤后神经源性异位骨化动物模型的研究已阐明了一系列关键的分子和细胞机制:首先,脊髓损伤通过去神经化作用加剧局部肌肉炎症,激活成骨微环境[33];其次,在神经源性异位骨化形成的早期阶段,神经信号和炎症信号起到驱动作用,而成骨细胞的成熟则更多依赖于后期的组织修复和微环境动态调节:如P物质通过神经炎症促进成骨分化[32];降钙素基因相关肽通过调控成纤维脂肪祖细胞的软骨分化加速异位骨形成[34];巨噬细胞分泌白细胞介素1β增强成纤维脂肪祖细胞的成骨基因表达[35];抑瘤素M通过Janus激酶/信号转导和转录活化因子信号驱动成骨分化[2];最后,巨噬细胞和成纤维脂肪祖细胞在脊髓损伤后神经源性异位骨化模型中具有核心作用,巨噬细胞耗竭将减少神经源性异位骨化体积90%,而成纤维脂肪祖细胞是神经源性异位骨化的主要成骨前体细胞来 源[32,36]。 尽管脊髓损伤后神经源性异位骨化模型在模拟神经源性异位骨化病理特征方面取得了一定进展,但仍存在一些局限:多数模型依赖化学性肌肉损伤(如心脏毒素注射),与人类损伤机制存在差异;不同模型异质性受动物品系、性别、年龄及实验条件的影响[5-6];此外,当前神经源性异位骨化模型缺乏系统的康复相关评估体系,限制了其临床转化价值。未来的研究需进一步优化实验设计,特别是整合多因素(如去神经、炎症、感染和代谢紊乱等)以更好地模拟临床神经源性异位骨化的复杂性,并丰富评价方法。表1系统总结了近年来脊髓损伤诱导神经源性异位骨化动物模型的研究设计、主要发现及相关分子机制[2,32-41]。 从表1归纳的研究可见,构建脊髓损伤诱导神经源性异位骨化动物模型的造模方法主要有以下几种:其一,脊髓损伤联合心脏毒素;其二,脊髓损伤联合去神经化与心脏毒素;其三,脊髓损伤联合肌腱损伤;其四,脊髓损伤联合心脏毒素与脂多糖。这几种方法都各有优势和局限,具体情况详见表2[2,32-33,35,40-41]。 2.2.2 创伤性脑损伤后神经源性异位骨化模型 创伤性脑损伤后神经源性异位骨化模型通过控制性皮质冲击联合外周创伤(如跟腱切断、股骨骨折)揭示了中枢神经损伤与局部组织病理的协同作用机制。研究表明,创伤性脑损伤不仅引发局部脑组织损伤,还可通过系统性神经内分泌信号(如降钙素基因相关肽、肾上腺素)和炎症级联反应重塑全身性微环境,显著增加神经源性异位骨化的发生率与严重程度[42]。创伤性脑损伤本身不足以独立诱导神经源性异位骨化,其与肌肉/骨骼损伤的协同效应是触发神经源性异位骨化的关键,具体机制涉及多个维度:在神经调控层面,创伤性脑损伤后脑源性降钙素基因相关肽通过降钙素受体样受体/降钙素受体辅助蛋白1受体激活Wnt/β-catenin通路,促进成纤维脂肪祖细胞向软骨细胞分化并启动软骨内骨化进程[34];在缺氧微环境层"
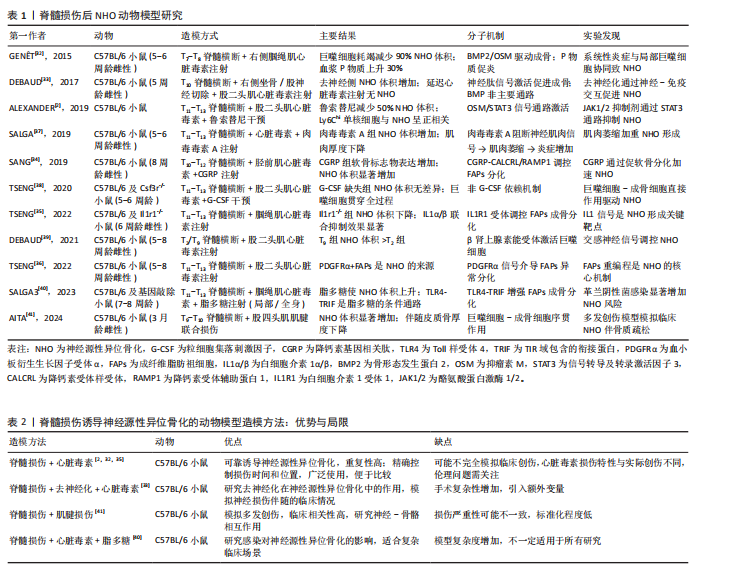
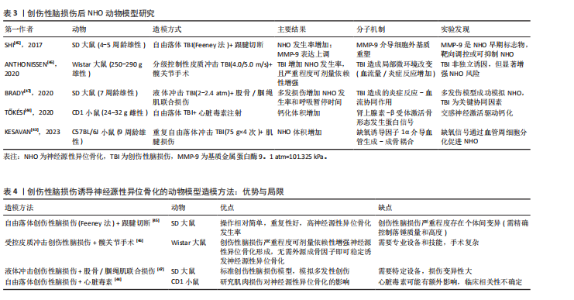
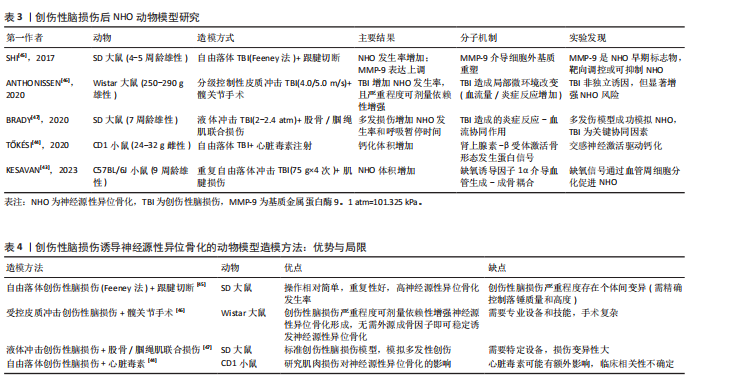
面,缺氧诱导因子1α信号通过上调血管内皮生长因子和促红细胞生成素的表达,促进血管周细胞向成骨细胞分化并加速矿化[43];在交感神经层面,肾上腺素通过β2-肾上腺素能受体增强巨噬细胞的抗炎极化特性,间接激活Runt相关转录因子2阳性成骨前体细胞的成骨潜能[44]。 未来研究应基于现有模型,深入探究创伤性脑损伤后神经源性异位骨化在多发创伤中的动态演变及其与其他损伤的交互机制;在转化医学层面,需着力开发精准靶向治疗策略,如焦磷酸盐、PHD2抑制剂等[43-44],以期有效降低神经源性异位骨化发病率,并显著改善患者的长期功能预后。表3系统总结了近年来创伤性脑损伤诱导神经源性异位骨化动物模型的研究设计、主要发现及相关分子机制[43-47]。 通过表3总结的研究内容可以发现,创伤性脑损伤诱导神经源性异位骨化的动物模型主要涵盖以下几种造模方式:其一是自由落体创伤性脑损伤联合跟腱切断;其二是受控皮质冲击创伤性脑损伤联合髋关节手术;其三是液体冲击创伤性脑损伤联合股骨/腘绳肌损伤;其四是自由落体创伤性脑损伤联合心脏毒素注射。这几种方法均存在着各自的优势与局限,见表4[44-47]。 2.3 成骨前体细胞在神经源性异位骨化中的核心作用 无论是脊髓损伤还是创伤性脑损伤后神经源性异位骨化动物模型,成骨前体细胞的募集、分化和功能异常激活均构成神经源性异位骨化形成的核心驱动因素。成骨前体细胞按照来源可分为局灶来源和迁徙来源。局灶来源的成骨前体细胞主要包括骨骼肌组织内的卫星细胞、成纤维脂肪祖细胞及血管周细胞,这些细胞在正常条件下主要参与肌肉的修复与再生;迁徙来源的成骨前体细胞则主要来自骨髓的骨髓间充质干细胞和神经内膜祖细胞。在脊髓损伤或创伤性脑损伤所诱导的病理环境影响下,这些成骨前体细胞的分化轨迹发生偏移,表现出异常的成骨潜能。 2.3.1 局灶来源的成骨前体细胞 (1)卫星细胞的功能异常:卫星细胞是位于骨骼肌基底膜与质膜间的组织特异性干细胞,通过自我更新和肌源性分化参与肌肉生长、损伤修复及再生调控[48-50]。在正常肌肉受损情况下,卫星细胞被激活、增殖和分化为肌原细胞,进而融合形成新的肌纤维以替代受损组织,维持肌肉的功能完整性[48-49]。在脊髓损伤病理环境中,卫星细胞的激活与分化能力遭受系统性抑制,表现为肌生成因子(生肌因子5/成肌分化蛋白)表达显著下调,同时局部微环境被炎性递质(如肿瘤坏死因子α、白细胞介素6)和纤维化信号(转化生长因子β)重塑,形成阻碍卫星细胞正常分化的分子屏障[48,51]。尽管脊髓损伤后卫星细胞仍具备分化为肌原细胞的潜能,但其分化产物无法有效修复肌纤维,且卫星细胞未直接参与成骨分化[48,52]。卫星细胞功能缺陷可能通过破坏与成纤维脂肪祖细胞的动态平衡间接推动神经源性异位骨化形成。研究显示,巨噬细胞等免疫细胞分泌的促炎因子(如白细胞介素1β)与生长因子(骨形态发生蛋白2)不仅与卫星细胞形成信号通路竞争,还通过抑制卫星细胞介导的肌再生能力,使卫星细胞对成纤维脂肪祖细胞的抑制作用减弱,促使成纤维脂肪祖细胞异常增殖及成骨分化[32]。综上,卫星细胞虽非神经源性异位骨化的直接细胞来源,但其功能障碍引发的肌再生失效与细胞互作失衡,共同构成了神经源性异位骨化形成的间接病理基础。 (2)成纤维脂肪祖细胞的成骨重编程:成纤维脂肪祖细胞是骨骼肌间质中的驻留间充质干细胞,在正常肌肉修复中,成纤维脂肪祖细胞通过精准调控实现损伤响应与稳态维持。在急性损伤时,成纤维脂肪祖细胞快速扩增以响应损伤信号,与巨噬细胞协同促进细胞外基质重塑及卫星细胞介导的肌再生,在修复后期,成纤维脂肪祖细胞通过程序性凋亡清除冗余细胞,从而防止组织纤维化[53-54];然而在脊髓损伤引发的神经源性异位骨化病理进程中,成纤维脂肪祖细胞经历病理性重编程,其增殖分化平衡被打破,最终转化为成骨前体细胞[32,36,51]。这种成骨重编程的转变在神经源性异位骨化中受多个因素影响,包括局部微环境的变化、炎症递质(如肿瘤坏死因子α、白细胞介素6)和纤维化信号(如转化生长因子β)的调控等。 首先,脊髓损伤后通过下调成骨抑制性miR-214-3p/miR-199a-3p削弱抑制信号,同时上调促成骨miR-20a-5p/miR-199a-5p维持持续激活状态,同时通过血小板衍生生长因子受体α信号通路的异常激活触发成纤维脂肪祖细胞表型转换,增强成纤维脂肪祖细胞的成骨潜能,最终促进神经源性异位骨化的发生[51];其次,中枢神经系统损伤后,局部组织中转化生长因子β过表达通过转化生长因子β受体Ⅰ/Ⅱ-Smad通路抑制肿瘤坏死因子介导的成纤维脂肪祖细胞凋亡,促使其向纤维化/成骨表型偏移,而脂多糖通过Toll样受体4/TIR域包含的衔接蛋白通路进一步放大其成骨潜能,表现为Runt相关转录因子2和碱性磷酸酶等基因的剂量依赖性上调及体外矿化能力增强[53];最后,降钙素基因相关肽含量增加,通过降钙素受体样受体/受体活性修饰蛋白1受体激活性别决定区Y框蛋白9和蛋白聚糖等软骨分化标志物,并与骨形态发生蛋白2形成协同效应,同时巨噬细胞分泌的抑瘤素M/白细胞介素1β通过"

Janus激酶-信号转导和转录活化因子/白细胞介素1受体通路增强成纤维脂肪祖细胞矿化能力[34]。 与创伤性脑损伤后神经源性异位骨化依赖骨形态发生蛋白信号不同,脊髓损伤后神经源性异位骨化呈现独特的骨形态发生蛋白非依赖性特征。TSENG等[35]研究表明骨形态发生蛋白抑制剂LDN-193189对骨化无显著影响,而GUEGUEN等[51]发现miR-214-3p介导的骨形态发生蛋白2间接调控可能仅在特定条件(如心脏毒素联合损伤)下发挥作用,这种机制的差异提示神经源性异位骨化存在病因特异性的分子开关。神经源性异位骨化病理性骨组织不仅包含成熟骨基质,还具备功能性造血生态位。研究表明,神经源性异位骨化的形成过程伴随巨噬细胞介导的血管化及免疫微环境构建,这为成纤维脂肪祖细胞的成骨分化提供了必要的生物学支撑[55]。成纤维脂肪祖细胞在神经源性异位骨化中的核心作用体现在中枢神经系统引发的信号传导失衡状态下,突破生理性调控阈值,通过表观重塑、炎症驱动及微环境互作完成向成骨前体细胞的不可逆转变,最终以软骨内骨化为主导模式推动异位骨化组织形成。 (3)血管周细胞的低氧响应:血管周细胞作为紧密包绕微血管基底膜的多潜能前体细胞,具有微环境依赖性,在低氧、炎症等应激条件下可被重编程为成骨前体细胞。创伤性脑损伤后神经源性异位骨化模型中,局部组织低氧通过激活缺氧诱导因子1α通路驱动血管周细胞活化,表现为损伤部位细胞密度显著升高及成骨标志物(胶原蛋白Ⅱ/Ⅹ型)表达上调,而脯氨酸羟化酶2抑制剂IOX2的应用可协同增强其成骨潜能,证实低氧信号是启动成骨转换的关键触发因素[43]。MATTHEWS等[56]研究表明,创伤性异位骨化病灶中超过50%的成骨/软骨细胞来源于α-平滑肌肌动蛋白阳性血管周细胞,其迁移与分化受骨形态发生蛋白2信号轴调控,且低氧刺激可显著增强其向病灶区的趋化能力。神经源性异位骨化病灶中还观察到低氧-炎症耦合特征,血管周细胞可能通过保守的缺氧诱导因子1α-骨形态发生蛋白信号轴在神经源性异位骨化中发挥作用,但现有证据主要基于动物实验,人类神经源性异位骨化样本中该细胞群体的空间分布、分子特征及其与神经损伤程度的关联仍需深入解析[43,56]。血管周细胞作为微环境敏感型成骨前体细胞,其低氧响应机制为理解神经源性异位骨化的细胞起源提供了新视角,但需通过临床转化研究验证病理贡献度及干预靶向性。 2.3.2 迁徙来源的成骨前体细胞 除了局部来源的成骨前体细胞外,来自远端组织的迁徙性成骨前体细胞也被认为是重要的神经源性异位骨化的细胞来源之一。此类细胞在神经源性异位骨化中的具体作用机制尚未完全阐明,少量研究提供了有关骨髓间充质干细胞和神经内膜祖细胞可能参与神经源性异位骨化形成的初步证据。 (1)骨髓间充质干细胞的成骨分化:骨髓间充质干细胞作为创伤性异位骨化的关键效应细胞,它的跨组织迁移与微环境响应机制为理解病理性成骨提供了证据。在手术诱导创伤性异位骨化模型中,SUDA等[57]鉴定出表达碱性磷酸酶/Ⅰ型胶原蛋白的循环性成骨前体细胞亚群,这类细胞借助血液循环迁移到损伤部位,在骨形态发生蛋白2等骨诱导因子驱动下分化为成熟成骨细胞,这首次指出骨髓源性细胞对远端骨化的贡献。CHEN等[58]发现,在缺氧条件下,缺氧诱导因子1α上调可促进Smad1/5/8磷酸化,进而激活骨钙素和骨桥蛋白等成骨基因,使骨髓间充质干细胞在异位部位形成钙化结构。骨髓间充质干细胞的成骨分化受到抑瘤素M、白细胞介素1β、骨形态发生蛋白2等多因子调控,其归巢行为通过CXC趋化因子受体4/基质细胞衍生因子1等信号轴实现,抑制该过程可显著降低软组织骨化程度[59-60]。在创伤性异位骨化小鼠模型中,研究显示异位骨组织周围的腘绳肌中可分离出骨髓间充质干细胞,并且显示出有一定的成骨潜能,这表明骨髓间充质干细胞的成骨分化直接影响创伤性异位骨化的发生发展[32,61]。 然而,骨髓间充质干细胞是否参与神经源性异位骨化的发病尚不清楚。此外,神经损伤特有的神经-免疫交互(如降钙素基因相关肽释放或交感神经活化)是否增强骨髓间充质干细胞归巢和成骨分化仍待进一步验证。 (2)神经内膜祖细胞的迁移:神经内膜祖细胞被认为是神经源性异位骨化的潜在成骨前体细胞来源之一。LAZARD等[62]首次报道通过骨形态发生蛋白2诱导创伤性异位骨化模型,神经内膜中claudin-5阳性祖细胞具有多谱系分化潜能,这些细胞可在三四天后通过外渗离开循环系统,迁移到新生骨部位并参与创伤性异位骨化的形成,其迁移路径为:神经内膜祖细胞从神经中脱离,通过血管运输至骨形成部位,并通过外渗进入目标位点。这一研究揭示了神经微环境与骨化病灶的信号传递路径,表明中枢神经损伤可能通过释放驻留祖细胞,在远端软组织内重编程为功能性成骨细胞。这种机制与经典骨髓源性或间质源性成骨前体细胞形成互补,为神经源性异位骨化的多细胞起源理论提供了实验支撑,但仍需进一步验证。 2.4 局部组织微环境对神经源性异位骨化的影响 局部组织成骨微环境作为神经源性异位骨化发生的关键调控因素之一,主要覆盖了缺氧微环境、新血管生成微环境以及炎症微环境等方面,这些微环境因素借助彼此间的相互作用以及协同调控机制,对成骨前体细胞的增殖、迁移以及成骨分化进程产生影响,推动神经源性异位骨化的发生与发展。在神经源性异位骨化中,脊髓损伤或创伤性脑损伤引发的全身性和局部病理变化进一步放大了这些微环境的复杂性和活性。 2.4.1 缺氧微环境 缺氧微环境是神经源性异位骨化的关键驱动因素之一,借助缺氧诱导因子1α构建的多层级调控网络,在成骨分化、软骨模板搭建以及血管生成方面有着系统性作用。首先,缺氧诱导因子1α通过骨形态发生蛋白信号轴直接激活成骨分化程序。缺氧诱导因子1α协同骨形态发生蛋白配体(如骨形态发生蛋白4)增强Ⅰ型胶原蛋白/骨钙素等骨基质基因表达,从而提高骨髓间充质干细胞的矿化能力。该过程还依赖Smad1/5/8-Runt相关转录因子2/碱性磷酸酶信号轴的激活,而抑制缺氧诱导因子1α可显著降低创伤性异位骨化的发生率[58,63-64]。其次,DUVAL等[65]发现,在低氧情况下,缺氧诱导因子1α可上调Sox9/蛋白聚糖等软骨分化标志物,并通过增强糖酵解通量为后续软骨向骨转换提供代谢基础。这表明缺氧诱导因子1α通过代谢重编程与表观调控介导软骨内骨化。 研究还显示,缺氧诱导因子1α通过上调血管内皮生长因子促进新生血管形成,而血管周细胞在低氧-炎症微环境中通过骨形态发生蛋白/血管内皮生长因子增强成骨分化,提示缺氧诱导因子1α在血管化与成骨过程中具有协同效应[43]。此外,中枢神经系统缺氧还与炎症信号产生协同放大效应。创伤性脑损伤小鼠血清显著促进血管周细胞增殖和分化,提示缺氧诱导因子1α可能与白细胞介素1β/肿瘤坏死因子α等炎症因子形成交叉调控,增强成骨前体细胞成骨潜能,促进神经源性异位骨化进展[43]。与经典创伤性异位骨化模型中由急性创伤性缺血引发的缺氧不同,神经源性异位骨化的低氧状态常与神经源性血管舒缩异常及慢性炎症引发的缺氧诱导因子1α持续激活有关,针对缺氧诱导因子1α的干预策略需考虑神经源性异位骨化的特异性。 2.4.2 血管生成微环境 新血管生成是神经源性异位骨化病理进程中的关键微环境事件,通过“血管-成骨偶联”机制发挥作用。首先,新生血管网络为骨化给予代谢支撑,病理性血管重塑为成骨前体细胞提供迁移通道。DILLING等[66]通过骨形态发生蛋白2诱导创伤性异位骨化模型证明血管生成比软骨模板出现得早(提前48-72 h),其空间分布与骨化进程有严格先后顺序。此外,血管内皮细胞的定向迁移是神经源性异位骨化启动的必要条件,阻断血管生成能让神经源性异位骨化发生率下降70%以上[67]。GIRARD等[55]进一步揭示,神经源性异位骨化病灶中异常增生的血管内皮表达高水平的细胞间黏附分子1/血管细胞黏附分子1,这些分子介导循环性成骨前体细胞的跨内皮迁移,使病灶区成骨前体细胞密度增加。 血管内皮生长因子可促进成骨前体细胞的增殖、归巢和成骨分化,同时抑制其成脂分化。由血管内皮生长因子受体1/2介导的经典促血管生成效应通过增加局部血供,促进成骨前体细胞增殖和归巢[68]。血管内皮生长因子与骨形态发生蛋白2构成协同放大回路,骨形态发生蛋白2通过上调血管内皮生长因子的表达促进血管新生,同时增强成骨前体细胞的归巢和成骨分化,加速软骨内骨化的进程[68]。如果血管内皮生长因子缺失,会致使Runt相关转录因子2表达下调,并且提高过氧化物酶体增殖物激活受体γ介导的骨髓间充质干细胞成脂分化[69]。总体而言,血管生成微环境为成骨前体细胞提供代谢支持、促进成骨前体细胞迁移和分化,推动了神经源性异位骨化进程。 2.4.3 炎症微环境 炎症微环境是神经源性异位骨化发生与发展的核心驱动力,其中巨噬细胞作为免疫调节枢纽,通过分泌促炎因子、调控成骨前体细胞分化,在神经源性异位骨化的炎症启动与成骨进程中发挥作用。 首先,脊髓损伤后巨噬细胞分泌白细胞介素1β,通过白细胞介素1受体1信号显著增强成纤维脂肪祖细胞中Runt相关转录因子2和Ⅰ型胶原α1等成骨基因表达。研究表明,白细胞介素1受体1敲除或白细胞介素1α/β中和抗体干预可减少神经源性异位骨化体积[35]。此外,巨噬细胞源性抑瘤素M通过抑瘤素M受体:糖蛋白130受体复合体激活Janus激酶1/2-信号转导和转录活化因子3通路,驱动成纤维脂肪祖细胞成骨转分化及骨基质矿化,而Janus激酶1/2抑制剂可抑制此过程[2,70]。GENêT等[32]通过巨噬细胞清除使脊髓损伤后神经源性异位骨化体积减少90%,显著高于P物质的抑制效果,提示巨噬细胞的作用超越单一促炎因子或神经递质。 巨噬细胞的极化方向也会对神经源性异位骨化产生巨大差异。早期M1型巨噬细胞通过白细胞介素1β、肿瘤坏死因子α等促炎因子激活骨形态发生蛋白信号及缺氧诱导因子1α,促进成骨基因表达,其作用可被P物质等神经肽进一步放大[63,71]。同时,M1型巨噬细胞分泌的血管内皮生长因子虽支持血管生成,但中枢神经系统损伤后诱导的神经炎症持续性促进M1型巨噬细胞活化导致炎症信号过度扩增,成为神经源性异位骨化成骨功能上调的关键诱因[63,72]。巨噬细胞M1极化可分泌CXC趋化因子12并促进其与间充质干细胞表面CXC趋化因子受体4结合,从而使间充质干细胞定向分化[73]。相比之下,M2型巨噬细胞在后期通过转化生长因子β1、白细胞介素10等抗炎因子缓解炎症,并经由转化生长因子β1-Smad2/3通路及血管内皮生长因子稳态释放支持骨修复与血管成熟[74-76]。因此,通过调控巨噬细胞的分化方向可能也是未来防治神经源性异位骨化的重要方式。 2.5 神经系统对神经源性异位骨化的调控机制 中枢神经系统损伤后,神经因子在神经源性异位骨化形成中的调控作用日益受到关注,不仅参与神经修复,还通过影响成骨细胞的分化与活性、调节细胞因子网络,在神经源性异位骨化的病理生理过程中发挥核心作用。其中神经生长因子、降钙素基因相关肽和P物质在神经源性异位骨化形成中表现出尤为突出的效应。 2.5.1 神经因子(神经生长因子、降钙素基因相关肽、P物质)的促骨化作用 (1)神经生长因子:神经生长因子在中枢神经系统损伤后可由受损神经纤维、脑脊液渗漏及局部细胞(如平滑肌细胞、巨噬细胞等)释放,并激活酪氨酸激酶A受体,启动多条下游信号通路[68,77-78]。神经生长因子/酪氨酸激酶A复合物磷酸化后,主要通过磷酯酶Cγ、磷脂酰肌醇3激酶/蛋白激酶B、丝裂原活化蛋白激酶激酶/细胞外调节蛋白激酶3条经典通路发挥作用[79-80]。其中,磷酯酶Cγ可诱导内质网Ca2?释放并激活磷酯酶C;而磷脂酰肌醇3激酶/蛋白激酶B及丝裂原活化蛋白激酶激酶/细胞外调节蛋白激酶则协同磷酸化环磷腺苷效应元件结合蛋白,进一步激活核因子κB[80-81],从而诱导骨形态发生蛋白2的表达并激活Smads信号,推动成骨前体细胞向成骨方向分化,同时上调Runt相关转录因子2、Osterix及碱性磷酸酶等骨标志物的表达[68,77-78]。此外,神经生长因子通过酪氨酸激酶A/丝裂原活化蛋白激酶激酶/细胞外调节蛋白激酶通路刺激血管周细胞及感觉神经末梢释放降钙素基因相关肽,增强病灶处神经血管侵入,诱导间充质干细胞通过转化生长因子β/Smad2/3通路分化为软骨/成骨细胞,并启动创伤性异位骨化进程[82-83]。研究还发现,神经生长因子能够激活肥大细胞去颗粒化,释放组胺、转化生长因子β等促骨因子,进一步招募间充质干细胞并诱导其分化为软骨或成骨细胞[84]。动物实验证实,使用酪氨酸激酶A抑制剂AR786处理后,微管蛋白β3阳性神经纤维密度显著降低(约85%),软骨相关基因Sox9与Ⅱ型胶原表达也明显下调[83]。神经生长因子还可通过酪氨酸激酶A/磷脂酰肌醇3激酶/蛋白激酶B/缺氧诱导因子1α通路上调血管内皮生长因子表达,促进血管生成,为成骨提供氧气支持[85-86]。但该机制目前主要基于创伤性脑损伤相关神经源性异位骨化研究,其在脊髓损伤后神经源性异位骨化中的特异性尚需进一步验证[68,87]。基于上述研究,针对神经生长因子-酪氨酸激酶A信号进行抑制可减少神经入侵及骨化体积,但需权衡它对神经修复的潜在负面影响[82-83]。 (2)降钙素基因相关肽:在中枢神经系统损伤后,降钙素基因相关肽通过感觉神经释放或血脑屏障渗漏进入外周,经降钙素受体样受体/受体活性修饰蛋白1激活多条信号通路[34,88-89]。降钙素基因相关肽与降钙素受体样受体/受体活性修饰蛋白1结合后,一方面通过Gαs-环磷酸腺苷–蛋白激酶A通路上调胞内环磷酸腺苷水平并激活蛋白激酶A[90];另一方面,它可以偶联Gαq-磷酯酶C-IP?/Ca2?通路,引发胞内Ca2?释放,从而激活钙/钙调蛋白依赖性蛋白激酶Ⅱ与蛋白激酶C,增强Wnt/β-catenin信号[91]。这两条信号通路共同促进Sox9、Runt相关转录因子2等软骨/成骨相关基因的表达,推动软骨的分化与矿化[34,88,92]。 体内外研究均表明,降钙素基因相关肽通过其受体(降钙素受体样受体/受体活性修饰蛋白1磷酯酶)作用于肌肉成纤维脂肪祖细胞,诱导其表达Sox9、蛋白聚糖等软骨标志物,促进其向软骨/成骨方向转化[34,93],此外,降钙素基因相关肽通过Gαs-环磷酸腺苷-蛋白激酶A通路抑制M1型巨噬细胞分泌肿瘤坏死因子α等炎症因子,从而缓解早期的炎症反应[94-95],同时可激活Hippo-Yes相关蛋白通路,促进M2型巨噬细胞极化,并对其成骨因子分泌呈现时间依赖性调控:损伤1-3 d内抑制骨形态发生蛋白2、骨形态发生蛋白6、Wnt10b与抑瘤素M表达,5-7 d则显著上调上述因子,进而增强成骨前体细胞中Sox9、Runt相关转录因子2与Osterix表达,促进软骨分化与骨形成[34,96]。 此外,降钙素基因相关肽还作为血管生成因子,通过Gαs–环磷酸腺苷-蛋白激酶A通路上调血管内皮生长因子/血管内皮生长因子受体2表达,促进新生血管的形成[97]。降钙素基因相关肽的缺失会导致血管内皮生长因子水平下降,从而抑制血管形成,延缓骨组织的修复与重塑[98-99]。此外,降钙素基因相关肽还直接作用于血管内皮细胞,促进一氧化氮合酶表达和一氧化氮释放,增强血管腔形成能力[100]。综上,降钙素基因相关肽具有作为神经源性异位骨化治疗靶点的潜力,通过靶向药物或受体拮抗剂,可能在特定病理阶段干预神经源性异位骨化的进程,从而治疗神经源性异位骨化。 (3)P物质:P物质是感觉神经释放的关键神经肽,通过神经源性炎症与骨代谢调控参与神经源性异位骨化的病理进程。中枢神经系统损伤后可诱导血清P物质水平显著升高,其表达强度与神经炎症微环境及损伤严重程度呈正相关[32,101-102]。P物质与神经激肽1受体结合,激活核因子κB和p38丝裂原活化蛋白激酶以及细胞外调节蛋白激酶1/2等关键信号通路,驱动巨噬细胞等免疫细胞释放肿瘤坏死因子α、白细胞介素1β及白细胞介素6等促炎因子,放大炎症反应,为神经源性异位骨化病灶的早期炎症浸润提供病理基础[32,103-104]。另外,P物质还可促进巨噬细胞M1极化,通过上调巨噬细胞内炎症相关基因表达,不仅增强其分泌炎症因子的能力,促进M1表型极化,还加剧组织破坏以及局部促骨化微环境的形成[71,105]。含有P物质的感觉神经直接影响骨代谢,推动骨吸收过程[106]。在神经源性异位骨化动物模型中,P物质-神经激肽1受体信号轴与骨形态发生蛋白通路协同激活,能驱动炎性反应,又能依靠骨形态发生蛋白2/Smad信号级联,诱导间充质干细胞向成骨谱系分化,加剧局部异位骨形成[107]。因此,通过对P物质及其下游通路进行干预可抑制神经源性异位骨化的发展。如神经激肽1受体拮抗剂RP-67580可显著抑制神经源性异位骨化体积及发生率[32,107]。 2.5.2 交感神经信号与β肾上腺素能受体通路 交感神经信号与β肾上腺素能受体通路在神经源性异位骨化的病理进程中发挥重要调控作用。首先,交感神经系统通过释放去甲肾上腺素靶向激活成骨细胞及骨髓间充质干细胞表面β肾上腺素能受体,抑制骨髓间充质干细胞的成脂分化倾向,并增强其成骨分化能力,从而重塑骨代谢平衡[55]。其次,交感神经系统通过调节骨髓微环境中CXC趋化因子12的分泌,直接影响间充质干细胞的归巢效率及免疫细胞的迁移,为神经源性异位骨化病灶提供细胞来源基础[39,108]。脊髓损伤后可释放儿茶酚胺,通过β肾上腺素能受体增强巨噬细胞抗炎特性,激活成骨前体细胞,从而促进神经源性异位骨化[39]。交感神经系统还通过增强血管内皮生长因子与缺氧诱导因子1α的协同效应,驱动病理性血管新生,为神经源性异位骨化病灶扩张提供营养支持[44]。因此,开发β受体阻滞剂(如普萘洛尔)或神经消融术等靶向干预方法可能对神经源性异位骨化产生防治作用。"

| [1] BALBONI TA, GOBEZIE R, MAMON HJ. Heterotopic ossification: Pathophysiology, clinical features, and the role of radiotherapy for prophylaxis. Int J Radiat Oncol Biol Phys. 2006;65(5):1289-1299. [2] ALEXANDER KA, TSENG HW, FLEMING W, et al. Inhibition of JAK1/2 Tyrosine Kinases Reduces Neurogenic Heterotopic Ossification After Spinal Cord Injury. Front Immunol. 2019;10:377. [3] RIZVI SMHA, SHARAF J, WILLIAMS KD, et al. Effectiveness of Prophylactic Interventions in Neurogenic Heterotopic Ossification (NHO): A Systematic Review. Cureus. 2022; 14(8):e27683. [4] VALBUENA VALECILLOS AD, GATER DR JR, et al. Concomitant Brain Injury and Spinal Cord Injury Management Strategies: A Narrative Review. J Pers Med. 2022;12(7): 1108. [5] AMPADIOTAKI MM, EVANGELOPOULOS DS, PALLIS D, et al. New Strategies in Neurogenic Heterotopic Ossification. Cureus. 2021;13(4):e14709. [6] 蔡童欣,雷敏聪,周逸钧,等.获得性异位骨化动物模型的系统综述[J].中国康复理论与实践,2022,28(4):429-438. [7] DING W, HU S, WANG P, et al. Spinal Cord Injury: The Global Incidence, Prevalence, and Disability From the Global Burden of Disease Study 2019. Spine (Phila Pa 1976). 2022;47(21):1532-1540. [8] RAWAT N, CHUGH S, ZACHARIAH K, et al. Incidence and characteristics of heterotopic ossification after spinal cord injury: a single institution study in India. Spinal Cord Ser Cases. 2019;5:72. [9] ALMANGOUR W, SCHNITZLER A, SALGA M, et al. Recurrence of heterotopic ossification after removal in patients with traumatic brain injury: A systematic review. Ann Phys Rehabil Med. 2016;59(4):263-269. [10] FRANZ S, RUST L, HEUTEHAUS L, et al. Impact of Heterotopic Ossification on Functional Recovery in Acute Spinal Cord Injury. Front Cell Neurosci. 2022;16:842090. [11] XIE Y, ZHANG J, JIN X, et al. Development and validation of a nomogram for predicting heterotopic ossification following spinal cord injury. Clin Neurol Neurosurg. 2024; 243:108348. [12] CITAK M, SUERO EM, BACKHAUS M, et al. Risk factors for heterotopic ossification in patients with spinal cord injury: a case-control study of 264 patients. Spine (Phila Pa 1976). 2012;37(23):1953-1957. [13] SULLIVAN MP, TORRES SJ, MEHTA S, et al. Heterotopic ossification after central nervous system trauma: A current review. Bone Joint Res. 2013;2(3):51-57. [14] LAL S, HAMILTON BB, HEINEMANN A, et al. Risk factors for heterotopic ossification in spinal cord injury. Arch Phys Med Rehabil. 1989;70(5):387-390. [15] JAZAYERI SB, BEYGI S, SHOKRANEH F, et al. Incidence of traumatic spinal cord injury worldwide: a systematic review. Eur Spine J. 2015;24(5):905-918. [16] JOHNSTON CB, DAGAR M. Osteoporosis in Older Adults. Med Clin North Am. 2020; 104(5):873-884. [17] VAN KUIJK AA, GEURTS AC, VAN KUPPEVELT HJ. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord. 2002; 40(7):313-326. [18] CONVENTE MR, WANG H, PIGNOLO RJ, et al. The immunological contribution to heterotopic ossification disorders. Curr Osteoporos Rep. 2015;13(2):116-124. [19] RANGANATHAN K, LODER S, AGARWAL S, et al. Heterotopic Ossification: Basic-Science Principles and Clinical Correlates. J Bone Joint Surg Am. 2015;97(13):1101-1111. [20] COELHO CV, BERALDO PS. Risk factors of heterotopic ossification in traumatic spinal cord injury. Arq Neuropsiquiatr. 2009;67(2B): 382-387. [21] REZNIK JE, BIROS E, MARSHALL R, et al. Prevalence and risk-factors of neurogenic heterotopic ossification in traumatic spinal cord and traumatic brain injured patients admitted to specialised units in Australia. J Musculoskelet Neuronal Interact. 2014; 14(1):19-28. [22] DIZDAR D, TIFTIK T, KARA M, et al. Risk factors for developing heterotopic ossification in patients with traumatic brain injury. Brain Inj. 2013;27(7-8):807-811. [23] BIERING-SØRENSEN B, KRISTENSEN IB, KJAER M, et al. Muscle after spinal cord injury. Muscle Nerve. 2009;40(4):499-519. [24] TAMBURIN S, FILIPPETTI M, MANTOVANI E, et al. Spasticity following brain and spinal cord injury: assessment and treatment. Curr Opin Neurol. 2022;35(6):728-740. [25] SANGARI S, PEREZ MA. Prevalence of spasticity in humans with spinal cord injury with different injury severity. J Neurophysiol. 2022;128(3):470-479. [26] BOEHL G, RAGUINDIN PF, VALIDO E, et al. Endocrinological and inflammatory markers in individuals with spinal cord injury: A systematic review and meta-analysis. Rev Endocr Metab Disord. 2022;23(5):1035-1050. [27] ZHANG C, JING Y, ZHANG W, et al. Dysbiosis of gut microbiota is associated with serum lipid profiles in male patients with chronic traumatic cervical spinal cord injury. Am J Transl Res. 2019;11(8):4817-4834. [28] ZAKRASEK EC, YURKIEWICZ SM, DIRLIKOV B, et al. Use of nonsteroidal anti-inflammatory drugs to prevent heterotopic ossification after spinal cord injury: a retrospective chart review. Spinal Cord. 2019;57(3):214-220. [29] 刘以俊,寇玉辉,张殿英,等.神经源性异位骨化发病机制的研究进展[J].中华创伤杂志,2020,36(9):853-858. [30] GENÊT F, JOURDAN C, LAUTRIDOU C, et al. The impact of preoperative hip heterotopic ossification extent on recurrence in patients with head and spinal cord injury: a case control study. PLoS One. 2011;6(8):e23129. [31] TALY AB, NAIR KP, JAYAKUMAR PN, et al. Neurogenic heterotopic ossification : a diagnostic and therapeutic challenge in neurorehabilitation. Neurol India. 2001;49(1): 37-40. [32] GENÊT F, KULINA I, VAQUETTE C, et al. Neurological heterotopic ossification following spinal cord injury is triggered by macrophage-mediated inflammation in muscle. J Pathol. 2015;236(2):229-240. [33] DEBAUD C, SALGA M, BEGOT L, et al. Peripheral denervation participates in heterotopic ossification in a spinal cord injury model. PLoS One. 2017;12(8): e0182454. [34] SANG X, WANG Z, SHI P, et al. CGRP accelerates the pathogenesis of neurological heterotopic ossification following spinal cord injury. Artif Cells Nanomed Biotechnol. 2019;47(1):2569-2574. [35] TSENG HW, KULINA I, GIRARD D, et al. Interleukin-1 Is Overexpressed in Injured Muscles Following Spinal Cord Injury and Promotes Neurogenic Heterotopic Ossification. J Bone Miner Res. 2022;37(3): 531-546. [36] TSENG HW, GIRARD D, ALEXANDER KA, et al. Spinal cord injury reprograms muscle fibroadipogenic progenitors to form heterotopic bones within muscles. Bone Res. 2022;10(1):22. [37] SALGA M, TSENG HW, ALEXANDER KA, et al. Blocking neuromuscular junctions with botulinum toxin A injection enhances neurological heterotopic ossification development after spinal cord injury in mice. Ann Phys Rehabil Med. 2019; 62(3):189-192. [38] TSENG HW, KULINA I, SALGA M, et al. Neurogenic Heterotopic Ossifications Develop Independently of Granulocyte Colony-Stimulating Factor and Neutrophils. J Bone Miner Res. 2020;35(11):2242-2251. [39] DEBAUD C, TSENG HW, CHEDIK M, et al. Local and Systemic Factors Drive Ectopic Osteogenesis in Regenerating Muscles of Spinal-Cord-Injured Mice in a Lesion-Level-Dependent Manner. J Neurotrauma. 2021;38(15):2162-2175. [40] SALGA M, SAMUEL SG, TSENG HW, et al. Bacterial Lipopolysaccharides Exacerbate Neurogenic Heterotopic Ossification Development. J Bone Miner Res. 2023;38(11): 1700-1717. [41] AITA R, UNNITHAN G, KLAYLAT T, et al. A novel mouse model of polytrauma with spinal cord injuryassociated heterotopic ossification. Eur Cell Mater. 2024;47:73-90. [42] SONG Y, BI L, ZHANG Z, et al. Increased levels of calcitonin gene-related peptide in serum accelerate fracture healing following traumatic brain injury. Mol Med Rep. 2012; 5(2):432-438. [43] KESAVAN C, GOMEZ GA, POURTEYMOOR S, et al. Development of an Animal Model for Traumatic Brain Injury Augmentation of Heterotopic Ossification in Response to Local Injury. Biomedicines. 2023;11(3):943. [44] TŐKÉSI N, KOZÁK E, FÜLÖP K, et al. Pyrophosphate therapy prevents trauma-induced calcification in the mouse model of neurogenic heterotopic ossification. J Cell Mol Med. 2020;24(20):11791-11799. [45] SHI WZ, JU JY, XIAO HJ, et al. Dynamics of MMP 9, MMP 2 and TIMP 1 in a rat model of brain injury combined with traumatic heterotopic ossification. Mol Med Rep. 2017; 15(4):2129-2135. [46] ANTHONISSEN J, STEFFEN CT, ALESSANDRI B, et al. Traumatic brain injury enhances the formation of heterotopic ossification around the hip: an animal model study. Arch Orthop Trauma Surg. 2020;140(8):1029-1035. [47] BRADY RD, ZHAO MZ, WONG KR, et al. A novel rat model of heterotopic ossification after polytrauma with traumatic brain injury. Bone. 2020;133:115263. [48] GÜNTHER S, KIM J, KOSTIN S, et al. Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell. 2013;13(5): 590-601. [49] SAMBASIVAN R, YAO R, KISSENPFENNIG A, et al. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138(17): 3647-3656. [50] COLLINS CA, OLSEN I, ZAMMIT PS, et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122(2):289-301. [51] GUEGUEN J, GIRARD D, RIVAL B, et al. Spinal cord injury dysregulates fibro-adipogenic progenitors miRNAs signaling to promote neurogenic heterotopic ossifications. Commun Biol. 2023;6(1):932. [52] MITCHELL KJ, PANNÉREC A, CADOT B, et al. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol. 2010;12(3):257-266. [53] LEMOS DR, BABAEIJANDAGHI F, LOW M, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med. 2015;21(7):786-794. [54] JOE AW, YI L, NATARAJAN A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010; 12(2):153-163. [55] GIRARD D, TOROSSIAN F, OBERLIN E, et al. Neurogenic Heterotopic Ossifications Recapitulate Hematopoietic Stem Cell Niche Development Within an Adult Osteogenic Muscle Environment. Front Cell Dev Biol. 2021;9:611842. [56] MATTHEWS BG, TORREGGIANI E, ROEDER E, et al. Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone. 2016;84:69-77. [57] SUDA RK, BILLINGS PC, EGAN KP, et al. Circulating osteogenic precursor cells in heterotopic bone formation. Stem Cells. 2009;27(9):2209-2219. [58] CHEN C, SONG C, LIU B, et al. Activation of BMP4/SMAD pathway by HIF-1α in hypoxic environment promotes osteogenic differentiation of BMSCs and leads to ectopic bone formation. Tissue Cell. 2024,88:102376. [59] LEES-SHEPARD JB, GOLDHAMER DJ. Stem cells and heterotopic ossification: Lessons from animal models. Bone. 2018;109:178-186. [60] PULIK Ł, MIERZEJEWSKI B, SIBILSKA A, et al. The role of miRNA and lncRNA in heterotopic ossification pathogenesis. Stem Cell Res Ther. 2022;13(1):523. [61] CHU DT, PHUONG TNT, TIEN NLB, et al. An Update on the Progress of Isolation, Culture, Storage, and Clinical Application of Human Bone Marrow Mesenchymal Stem/Stromal Cells. Int J Mol Sci. 2020;21(3):708. [62] LAZARD ZW, OLMSTED-DAVIS EA, SALISBURY EA, et al. Osteoblasts Have a Neural Origin in Heterotopic Ossification. Clin Orthop Relat Res. 2015;473(9):2790-2806. [63] WANG H, LINDBORG C, LOUNEV V, et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J Bone Miner Res. 2016;31(9):1652-1665. [64] AGARWAL S, LODER S, BROWNLEY C, et al. Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc Natl Acad Sci U S A. 2016; 113(3):E338-347. [65] DUVAL E, BAUGÉ C, ANDRIAMANALIJAONA R, et al. Molecular mechanism of hypoxia-induced chondrogenesis and its application in in vivo cartilage tissue engineering. Biomaterials. 2012;33(26):6042-6051. [66] DILLING CF, WADA AM, LAZARD ZW, et al. Vessel formation is induced prior to the appearance of cartilage in BMP-2-mediated heterotopic ossification. J Bone Miner Res. 2010;25(5):1147-1156. [67] YANG YQ, TAN YY, WONG R, et al. The role of vascular endothelial growth factor in ossification. Int J Oral Sci. 2012;4(2):64-68. [68] LI SN, RAN RY, CHEN J, et al. Angiogenesis in heterotopic ossification: From mechanisms to clinical significance. Life Sci. 2024;351:122779. [69] LIU Y, BERENDSEN AD, JIA S, et al. Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J Clin Invest. 2012;122(9): 3101-3113. [70] SIMS NA, LÉVESQUE JP. Oncostatin M: Dual Regulator of the Skeletal and Hematopoietic Systems. Curr Osteoporos Rep. 2024;22(1):80-95. [71] TUZMEN C, VERDELIS K, WEISS L, et al. Crosstalk between substance P and calcitonin gene-related peptide during heterotopic ossification in murine Achilles tendon. J Orthop Res. 2018;36(5):1444-1455. [72] SHI C, PAMER EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762-774. [73] ALBIERO M, PONCINA N, CICILIOT S, et al. Bone Marrow Macrophages Contribute to Diabetic Stem Cell Mobilopathy by Producing Oncostatin M. Diabetes. 2015; 64(8):2957-2968. [74] BARRUET E, MORALES BM, CAIN CJ, et al. NF-κB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI Insight. 2018;3(22):e122958. [75] HATSELL SJ, IDONE V, WOLKEN DM, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015;7(303):303ra137. [76] WANG X, LI F, XIE L, et al. Inhibition of overactive TGF-β attenuates progression of heterotopic ossification in mice. Nat Commun. 2018;9(1):551. [77] YANG X, MOU D, YU Q, et al. Nerve growth factor promotes osteogenic differentiation of MC3T3-E1 cells via BMP-2/Smads pathway. Ann Anat. 2022;239:151819. [78] BRACCI-LAUDIERO L, ALOE L, CAROLEO MC, et al. Endogenous NGF regulates CGRP expression in human monocytes, and affects HLA-DR and CD86 expression and IL-10 production. Blood. 2005;106(10):3507-3514. [79] MINNONE G, DE BENEDETTI F, BRACCI-LAUDIERO L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int J Mol Sci. 2017;18(5):1028. [80] REICHARDT LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545-1564. [81] HUANG EJ, REICHARDT LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609-642. [82] CHERIEF M, NEGRI S, QIN Q, et al. TrkA+ Neurons Induce Pathologic Regeneration After Soft Tissue Trauma. Stem Cells Transl Med. 2022;11(11):1165-1176. [83] LEE S, HWANG C, MARINI S, et al. NGF-TrkA signaling dictates neural ingrowth and aberrant osteochondral differentiation after soft tissue trauma. Nat Commun. 2021; 12(1):4939. [84] JIANG T, AO X, XIANG X, et al. Mast cell activation by NGF drives the formation of trauma-induced heterotopic ossification. JCI Insight. 2024;10(1):e179759. [85] NAKAMURA K, TAN F, LI Z, et al. NGF activation of TrkA induces vascular endothelial growth factor expression via induction of hypoxia-inducible factor-1α. Mol Cell Neurosci. 2011; 46(2):498-506. [86] LU QL, LIU J, ZHU XL, et al. Expression of nerve growth factor and hypoxia inducible factor-1α and its correlation with angiogenesis in non-small cell lung cancer. J Huazhong Univ Sci Technolog Med Sci. 2014;34(3):359-362. [87] ZHANG R, LIANG Y, WEI S. The expressions of NGF and VEGF in the fracture tissues are closely associated with accelerated clavicle fracture healing in patients with traumatic brain injury. Ther Clin Risk Manag. 2018;14:2315-2322. [88] ZHOU R, YUAN Z, LIU J, et al. Calcitonin gene-related peptide promotes the expression of osteoblastic genes and activates the WNT signal transduction pathway in bone marrow stromal stem cells. Mol Med Rep. 2016;13(6):4689-4696. [89] RUSSELL FA, KING R, SMILLIE SJ, et al. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014; 94(4):1099-1142. [90] WALKER CS, CONNER AC, POYNER DR, et al. Regulation of signal transduction by calcitonin gene-related peptide receptors. Trends Pharmacol Sci. 2010;31(10):476-483. [91] WANG Q, QIN H, DENG J, et al. Research Progress in Calcitonin Gene-Related Peptide and Bone Repair. Biomolecules. 2023;13(5): 838. [92] PEREIRA CT, ADAMS SH, LLOYD KCK, et al. Exploring the role of peripheral nerves in trauma-induced heterotopic ossification. JBMR Plus. 2024;9(1):ziae155. [93] WANG K, YAGHI OK, SPALLANZANI RG, et al. Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc Natl Acad Sci U S A. 2020;117(10):5402-5408. [94] FENG Y, TANG Y, GUO J, et al. Inhibition of LPS-induced TNF-alpha production by calcitonin gene-related peptide (CGRP) in cultured mouse peritoneal macrophages. Life Sci. 1997; 61(20):PL281-287. [95] DUAN JX, ZHOU Y, ZHOU AY, et al. Calcitonin gene-related peptide exerts anti-inflammatory property through regulating murine macrophages polarization in vitro. Mol Immunol. 2017;91:105-113. [96] ZHANG Q, WU B, YUAN Y, et al. CGRP-modulated M2 macrophages regulate osteogenesis of MC3T3-E1 via Yap1. Arch Biochem Biophys. 2021;697:108697. [97] XIONG J, WANG Z, BAI J, et al. Calcitonin gene-related peptide: a potential protective agent in cerebral ischemia-reperfusion injury. Front Neurosci. 2023;17:1184766. [98] MATSUI S, TANAKA M, KAMIYOSHI A, et al. Endogenous Calcitonin Gene-Related Peptide Deficiency Exacerbates Postoperative Lymphedema by Suppressing Lymphatic Capillary Formation and M2 Macrophage Accumulation. Am J Pathol. 2019;189(12):2487-2502. [99] XU J, WANG J, CHEN X, et al. The Effects of Calcitonin Gene-Related Peptide on Bone Homeostasis and Regeneration. Curr Osteoporos Rep. 2020;18(6):621-632. [100] SHI Z, WANG S, DENG J, et al. Neural Peptide α-CGRP Coregulated Angiogenesis and Osteogenesis via Promoting the Cross-Talk between Mesenchymal Stem Cells and Endothelial Cells. Biomed Res Int. 2022; 2022:1585840. [101] BRADY RD, SHULTZ SR, MCDONALD SJ, et al. Neurological heterotopic ossification: Current understanding and future directions. Bone. 2018;109:35-42. [102] LORENTE L, MARTÍN MM, ALMEIDA T, et al. Serum substance P levels are associated with severity and mortality in patients with severe traumatic brain injury. Crit Care. 2015; 19(1):192. [103] SALISBURY E, RODENBERG E, SONNET C, et al. Sensory nerve induced inflammation contributes to heterotopic ossification. J Cell Biochem. 2011;112(10):2748-2758. [104] SUN J, RAMNATH RD, ZHI L, et al. Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol. 2008;294(6):C1586-1596. [105] O’CONNOR TM, O’CONNELL J, O’BRIEN DI, et al. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201(2):167-180. [106] SHERMAN BE, CHOLE RA. A mechanism for sympathectomy-induced bone resorption in the middle ear. Otolaryngol Head Neck Surg. 1995;113(5):569-581. [107] KAN L, LOUNEV VY, PIGNOLO RJ, et al. Substance P signaling mediates BMP-dependent heterotopic ossification. J Cell Biochem. 2011;112(10):2759-2772. [108] FIELDING C, GARCÍA-GARCÍA A, KORN C, et al. Cholinergic signals preserve haematopoietic stem cell quiescence during regenerative haematopoiesis. Nat Commun. 2022;13(1):543. |
| [1] | Peng Hao, Jiang Yang, Song Yanping, Wu Quan, Yao Na, Chen Qigang, Shen Zhen. H-type angiogenesis and its role in various skeletal disease animal models [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(16): 4154-4165. |
| [2] | Wu Xiuli, Yan Xiaoxia, Ren Zhiqiang, Sun Nan, Li Jinju. Modeling methods and evaluation criteria in animal models of steroid-induced osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(21): 4560-4567. |
| [3] | Qian Zuping, Chen Yong, Ran Yan, Da Jingjing, Zha Yan. Diabetic nephropathy model: animal model, two-dimensional cell simulation and three-dimensional organoid model [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(17): 3632-3640. |
| [4] | Liu Chuang, Tan Longwang, Zhou Heshan, Zhang Chi. Adipose-derived mesenchymal stem cell exosomes for treating traumatic central nervous system injury [J]. Chinese Journal of Tissue Engineering Research, 2023, 27(19): 3061-3069. |
| [5] | Sun Xirao, Bao Jiaxin, Wang Chengyue. Construction of chitosan/mineralized collagen porous scaffold, osteogenic differentiation in vitro and biocompatibility [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(34): 5498-5503. |
| [6] | Wang Kang, Zhi Xiaodong, Wang Wei. Effect and mechanism of human amniotic epithelial cells on nerve injury repair [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(25): 4046-4051. |
| [7] | Chen Xianxiang, Liao Shijie, Li Boxiang, Li Chong, Huang Qian, Lin Chengsen, Liu Yun, Lu Rongbin, Ding Xiaofei. Characteristics and application on animal models of different species of Perthes disease [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(18): 2922-2929. |
| [8] | Wang Jing, Lu Changfeng, Peng Jiang, Zhu Chen, Xu Wenjing, Cheng Xiaoqing, Fang Jie, Zhu Yaqiong, Zhao Yanxu, Jiang Wen, Xu Hongguang, Wang Yu. Establishment and evaluation of traumatic neuroma model [J]. Chinese Journal of Tissue Engineering Research, 2020, 24(5): 716-719. |
| [9] | Xu Huijun, Zhang Mi, Shi Dongmei, Wu Saixuan, Lu Ying, Dong Ming, Niu Weidong. Fetal bovine serum exosomes promote the proliferation of osteoblasts [J]. Chinese Journal of Tissue Engineering Research, 2020, 24(31): 4961-4965. |
| [10] | Gu Jiangjiang, Zhao Fengchao, Cheng Qi, Tang Jinlong, Xu Shizhuang. Establishing a model of femoral head necrosis by alcohol burning in Cyan-Shank Partridge Chicken: a feasibility analysis [J]. Chinese Journal of Tissue Engineering Research, 2020, 24(17): 2700-2705. |
| [11] | Ge Yingjie1, Cai Miaoxin1, Li Linpeng1, Chen Haoyan1, Pang Zhihui2, Fan Yueguang3. Features of animal models of osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2019, 23(35): 5690-5696. |
| [12] | Wang Zirun, Xiao Chengwei, Hu Jiang. Construction of SD rat scoliosis model and the effect of asymmetric tension on bone structure development [J]. Chinese Journal of Tissue Engineering Research, 2019, 23(31): 5005-5009. |
| [13] | Jia Peng, Zhang Tao. Biological cage in intervertebral bone integration: histocytological properties of interface and healing mechanism of osseointegration [J]. Chinese Journal of Tissue Engineering Research, 2019, 23(26): 4249-4254. |
| [14] | Wu Yang-peng, Fan Xiao, Zhang Li. Establishment and evaluation of the animal model of acute spinal cord injury [J]. Chinese Journal of Tissue Engineering Research, 2016, 20(49): 7341-7348. |
| [15] | Li Jian-feng, Feng Shi-qing, Xia Run-fu, Yan Jin-yu. Design of an injury device to establish spinal cord dorsal compression injury models in rats [J]. Chinese Journal of Tissue Engineering Research, 2015, 19(18): 2856-2861. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||