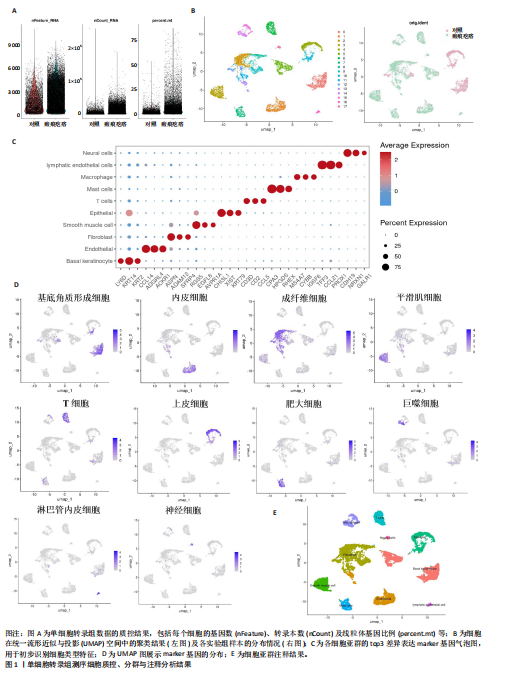
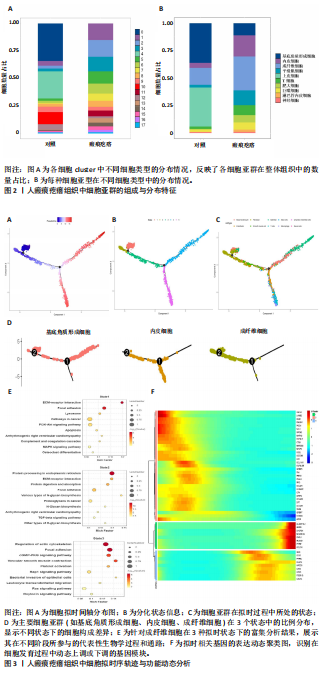
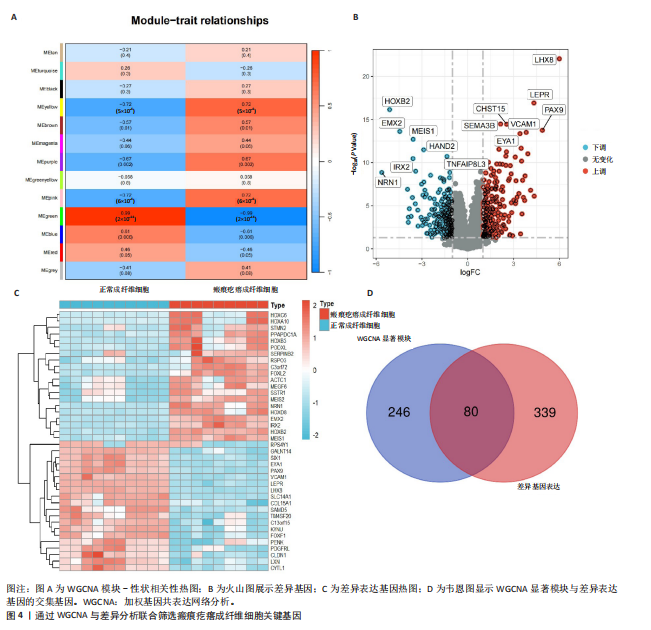
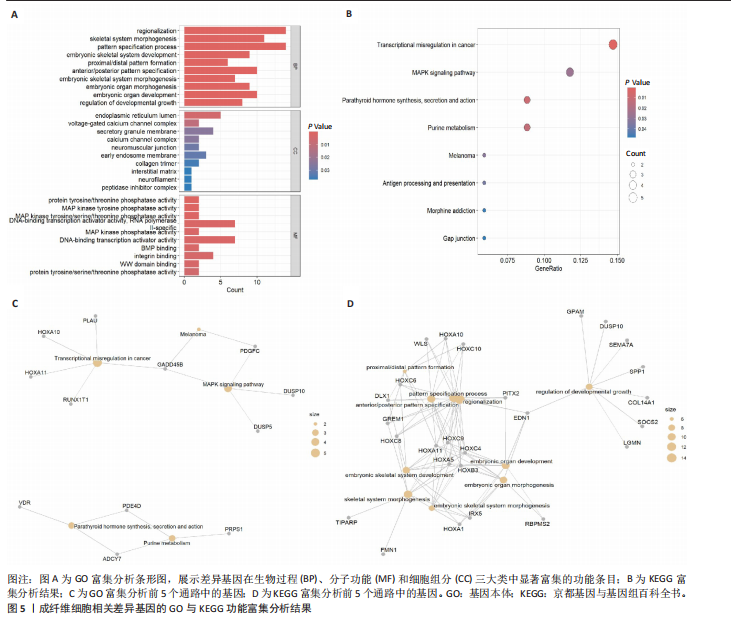
[1] HAWASH AA, INGRASCI G, NOURI K, et al. Pruritus in Keloid Scars: Mechanisms and Treatments. Acta Derm Venereol. 2021;101(10):adv00582.
[2] KELLY AP. Keloids. Dermatol Clin. 1988;6(3): 413-424.
[3] LEE SS, YOSIPOVITCH G, CHAN YH, et al. Pruritus, pain, and small nerve fiber function in keloids: a controlled study. J Am Acad Dermatol. 2004; 51(6):1002-1006.
[4] DOHI T, PADMANABHAN J, AKAISHI S, et al. The Interplay of Mechanical Stress, Strain, and Stiffness at the Keloid Periphery Correlates with Increased Caveolin-1/ROCK Signaling and Scar Progression. Plast Reconstr Surg. 2019;144(1):58e-67e.
[5] KASSI K, KOUAME K, KOUASSI A, et al. Quality of life in black African patients with keloid scars. Dermatol Reports. 2020;12(2):8312.
[6] FERNANDES MG, DA SILVA LP, CERQUEIRA MT, et al. Mechanomodulatory biomaterials prospects in scar prevention and treatment. Acta Biomater. 2022;150:22-33.
[7] WEI J, WANG Z, ZHONG C, et al. LncRNA MIR503HG promotes hypertrophic scar progression via miR-143-3p-mediated Smad3 expression. Wound Repair Regen. 2021;29(5): 792-800.
[8] BIAZUS SOARES G, MAHMOUD O, YOSIPOVITCH G. Pruritus in keloid scars: mechanisms and treatments. Ital J Dermatol Venerol. 2023;158(5):401-407.
[9] MUSKAT A, KOST Y, BALAZIC E, et al. Laser-Assisted Drug Delivery in the Treatment of Scars, Rhytids, and Melasma: A Comprehensive Review of the Literature. Aesthet Surg J. 2023;43(3):NP181-NP198.
[10] BAGABIR R, BYERS RJ, CHAUDHRY IH, et al. Site-specific immunophenotyping of keloid disease demonstrates immune upregulation and the presence of lymphoid aggregates. Br J Dermatol. 2012;167(5):1053-1066.
[11] SHARMA JR, LEBEKO M, KIDZERU EB, et al. In Vitro and Ex Vivo Models for Functional Testing of Therapeutic Anti-scarring Drug Targets in Keloids. Adv Wound Care (New Rochelle). 2019; 8(12):655-670.
[12] 李政宇.基于综合生物信息学筛选瘢痕疙瘩关键生物标志物及SDC4泛癌分析[D].太原:太原理工大学,2023.
[13] LV W, REN Y, HOU K, et al. Epigenetic modification mechanisms involved in keloid: current status and prospect. Clin Epigenetics. 2020;12(1):183.
[14] XIE R, LI C, ZHAO T, et al. Integration of Flow Cytometry and Single-Cell RNA Sequencing Analysis to Explore the Fibroblast Subpopulations in Keloid that Correlate with Recurrence. Adv Wound Care (New Rochelle). 2025. doi: 10.1089/wound.2024.0262.
[15] ZHONG C, SHI K, LI P, et al. Single-cell sequencing analysis and bulk-seq identify IGFBP6 and TNFAIP6 as novel differential diagnosis markers for postburn pathological scarring. Burns. 2024; 50(9):107255.
[16] SZKLARCZYK D, KIRSCH R, KOUTROULI M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638-D646.
[17] WEI W, LI Y, HUANG T. Using Machine Learning Methods to Study Colorectal Cancer Tumor Micro-Environment and Its Biomarkers. Int J Mol Sci. 2023;24(13):11133.
[18] UDDIN S, KHAN A, HOSSAIN ME, et al. Comparing different supervised machine learning algorithms for disease prediction. BMC Med Inform Decis Mak. 2019;19(1):281.
[19] MOTAMEDI F, PÉREZ-SÁNCHEZ H, MEHRIDEHNAVI A, et al. Accelerating Big Data Analysis through LASSO-Random Forest Algorithm in QSAR Studies. Bioinformatics. 2022;38(2):469-475.
[20] BIJL JJ, RIEGER E, VAN OOSTVEEN JW, et al. HOXC4, HOXC5, and HOXC6 expression in primary cutaneous lymphoid lesions. High expression of HOXC5 in anaplastic large-cell lymphomas. Am J Pathol. 1997;151(4):1067-1074.
[21] WU S, CHEN H, ZUO L, et al. Suppression of long noncoding RNA MALAT1 inhibits the development of uveal melanoma via microRNA-608-mediated inhibition of HOXC4. Am J Physiol Cell Physiol. 2020;318(5):C903-C912.
[22] ZHANG H, HAN B, TIAN S, et al. HOXC4 promotes proliferation of pancreatic cancer cells by increasing LDHA-mediated glycolysis. Aging (Albany NY). 2024;16(13):11103-11116.
[23] HAHN JM, MCFARLAND KL, COMBS KA, et al. Analysis of HOX gene expression and the effects of HOXA9 overexpression in fibroblasts derived from keloid lesions and normal skin. Wound Repair Regen. 2021;29(5):777-791.
[24] HAHN JM, GLASER K, MCFARLAND KL, et al. Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013;21(4):530-544.
[25] ZHANG N, GUO F, SONG Y. HOXC8/TGF-β1 positive feedback loop promotes liver fibrosis and hepatic stellate cell activation via activating Smad2/Smad3 signaling. Biochem Biophys Res Commun. 2023;662:39-46.
[26] XIAO J, LI Y, LIU Y, et al. The involvement of homeobox-C 4 in predicting prognosis and unraveling immune landscape across multiple cancers via integrated analysis. Front Genet. 2022;13:1021473.
[27] KIRK T, AHMED A, ROGNONI E. Fibroblast Memory in Development, Homeostasis and Disease. Cells. 2021;10(11):2840.
[28] JIAO H, FAN J, CAI J, et al. Analysis of Characteristics Similar to Autoimmune Disease in Keloid Patients. Aesthetic Plast Surg. 2015;39(5):818-825.
[29] WU J, DEL DUCA E, ESPINO M, et al. RNA Sequencing Keloid Transcriptome Associates Keloids With Th2, Th1, Th17/Th22, and JAK3-Skewing. Front Immunol. 2020;11:597741.
[30] LEE CC, TSAI CH, CHEN CH, et al. An updated review of the immunological mechanisms of keloid scars. Front Immunol. 2023;14:1117630.
[31] MARTIN CW, MUIR IF. The role of lymphocytes in wound healing. Br J Plast Surg. 1990;43(6):655-662.
[32] LI X, AN T, YANG Y, et al. TLR9 activation in large wound induces tissue repair and hair follicle regeneration via γδT cells. Cell Death Dis. 2024;15(8):598.
[33] XIA Y, WANG Y, SHAN M, et al. Advances in the pathogenesis and clinical application prospects of tumor biomolecules in keloid. Burns Trauma. 2022;10:tkac025.
[34] DENG CC, XU XY, ZHANG Y, et al. Single-cell RNA-seq reveals immune cell heterogeneity and increased Th17 cells in human fibrotic skin diseases. Front Immunol. 2025;15: 1522076.
[35] ZHANG X, WU X, LI D. The Communication from Immune Cells to the Fibroblasts in Keloids: Implications for Immunotherapy. Int J Mol Sci. 2023;24(20):15475.
[36] SHAN M, WANG Y. Viewing keloids within the immune microenvironment. Am J Transl Res. 2022;14(2):718-727.
[37] LI Y, LI M, QU C, et al. The Polygenic Map of Keloid Fibroblasts Reveals Fibrosis-Associated Gene Alterations in Inflammation and Immune Responses. Front Immunol. 2022;12:810290.
|