Chinese Journal of Tissue Engineering Research
Architecture of network medicine and its research progress
Li Ying1, Huang Wen-cong2
- 1Guangxi Colleges and Universities Key Laboratory of Scientific Computing &
Intelligent Information Processing of Guangxi Teachers Education University, Nanning 530001, Guangxi Zhuang Autonomous Region, China; 2School of Sports of Guangxi Teachers Education University, Nanning 530001,Guangxi Zhuang Autonomous Region, China
-
Received:2016-12-12Online:2017-04-28Published:2017-05-16 -
About author:Li Ying, Ph.D., Associate professor, Guangxi Colleges and Universities Key Laboratory of Scientific Computing & Intelligent Information Processing of Guangxi Teachers Education University, Nanning 530001, Guangxi Zhuang Autonomous Region, China -
Supported by:the Scientific Research Foundation of the Higher Education Institutions of Guangxi Zhuang Autonomous Region, China, No. KY2015YB188
CLC Number:
Cite this article
Li Ying1, Huang Wen-cong2. Architecture of network medicine and its research progress[J]. Chinese Journal of Tissue Engineering Research, doi: 10.3969/j.issn.2095-4344.2017.12.026.
share this article
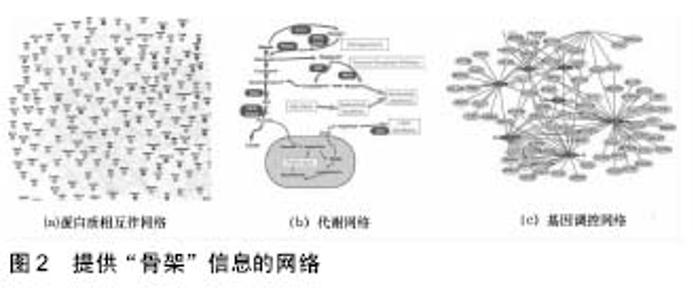
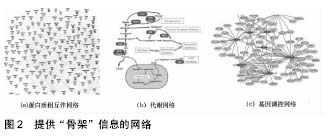
2.1 分子相互作用网络(Interactome) “Interactome”术语由一组以Bernard Jacq为首的法国科学家在1999年创造,指的是分子间的相互作用[5]。分子相互作用在属于不同生物化学家族(蛋白质、核酸、脂类、碳水化合物等)的分子之间产生,也可以在某个给定家族中产生[6]。分子相互作用网络在数学上一般表现为图,这些图通过参与其中的化合物的性质来分类[6]。分子相互作用网络中,节点是代谢物和诸如蛋白质、RNA分子和基因序列这样的分子,而边是可以用大量技术来识别的物理、生物化学和功能相互作用[7]。代谢网络、蛋白质-蛋白质相互作用网络和基因调控网络提供了关于细胞系统的关键的“骨架”信息,在这个“骨架”上还可以添加功能连接的额外层来调整生物的现实表现[7]。这些功能链接的网络代表一类细胞网络,它们可以间接或“概念的”得到,包括转录谱网络、表型谱网络、基因相互作用网络等。把提供“骨架”信息的网络和功能连接的网络整合起来就具备了强大的预测价值[8]。可以使用分子相互作用网络来发现疾病、对疾病进行分类及开发治疗方法,还可以用来预测疾病基因及识别疾病模块。 构造分子相互作用网络的方法有3种[7]。第一种是文献法,即通过编辑或综合处理文献中早已存在的数据,这些数据从一种或几种物理或生物化学相互作用中获得。第二种是计算法,即基于可用的“正交”信息的计算预测,例如序列相似性、基因顺序保存、完整序列基因的共现和共缺失及蛋白质结构信息。第3种是实验法,即应用于整个基因组或蛋白质组规模的系统的、无偏的高通量实验绘制策略。这些方法是互补的,但对所获得的网络图的解释是非常不同的。通过文献法得到的图展现使用可用信息的优点,但受限于出版数据的固有易变性限制,缺乏系统性及缺乏负面数据的报告。通过计算预测得到图是快速和有效的,通常包含大量节点和边,但这些图因其使用间接数据而必然是不完美的。通过高通量实验得到的图描述无偏的、系统的和控制良好的数据,但初始建图是困难的。下面是目前已经构建的各种分子相互作用网络。 2.1.1 提供“骨架”信息的网络及其模块性质 蛋白质相互作用网(Protein-Protein Interaction Networks):在蛋白质-蛋白质相互作用网中,节点代表蛋白质,边代表2种蛋白质之间的物理相互作用。边是无向的,因为不能说是哪种蛋白质结合了另一种蛋白质,即哪个伙伴从功能上影响另一个。在多种绘制蛋白质-蛋白质相互作用的方法中,有2种现在被广泛用于大规模绘制[7]。绘制二元相互作用主要是通过不断提高酵母双杂交(Y2H)系统的变异来进行的。蛋白质复合体成员的绘制,是通过亲和或免疫纯化来分离蛋白质复合体,随后是某种形式的质谱分析(AP/MS)来识别这些复合体的蛋白质构成[9-10]。Y2H数据集主要是包含直接的二元相互作用,而AP/MS复合物数据集由直接相互作用和数量上占优势的间接关联组成。通过高通量AP/MS来全面绘制共复合体的成员最初在酵母中进行,此后迅速进步为不断提高质量及完整性。由于技术原因,未来全面的AP/MS方法将专注于诸如酵母这样的单细胞和支原体[11],而Y2H方法能更快实现于复杂的多细胞生物[12]。使用Y2H技术来生成相互作用图始于酿酒酵母、线虫和果蝇等模型生物,最终包括人类的蛋白质相互作用图。从2005年到现在,人类蛋白质相互作用网不断得到完善[13-17]。 在蛋白质相互作用网图的早期实现中,由于准确性得不到保证,绘制系统的及全面的相互作用网的努力遭到怀疑。最近普及的蛋白质相互作用网图的一个实证框架允许估计使用高通量绘制方法的整体准确性和灵敏度[7]。使用这个框架需要估计4个关键参数[7]:①完整性:在一个给定搜索空间实际测试的物理蛋白质对的数量;②检测灵敏度:通过特定检测,哪些相互作用可以/不可以被发现;③采样灵敏度:任何相互作用检测的单一实施所发现的可检测相互作用的部分;④精确性:真实生物物理相互作用的比率。这些参数的详细考虑提供了特定高通量相互作用图的完整性和准确性的量化思想[18],在使用标准框架参数的情况下还允许比较多个图。相比之下,对文献来源数据库中可用的小规模实验结果比较多个图是不可能的,因为无法控制精确度、重现性和灵敏度。这个二元相互作用的实证框架提供了一种方法来估计蛋白质相互作用网络的大小,这反过来对定义了一个路线图以完成任何物种的蛋白质相互作用网图的绘制是必要的,类似的实证框架可以应用到更广泛地绘制其他类型的相互作用网络[19]。 蛋白质相互作用网络主要应用表现在以下几个方面:用于辅助识别疾病基因和枢纽基因[20-23];用于辅助识别特定疾病的致病基因参与的途径[24];用于识别药物靶标[25];用于识别兼职功能蛋白质[26];用于识别蛋白质功能模块[27];用来研究特定疾病蛋白质的网络拓扑[17];用于研究蛋白质相互作用网络的演化等[28]。 代谢网络(Metabolic Network):代谢网络综合描述一种特定细胞或生物的所有可能生物化学反应[29-30]。代谢网络的节点是生物化学代谢物,边是把一种代谢物转换成另一种代谢物的反应,或者是催化这些反应的酶[30-31]。边的有向或无向取决于反应是否可逆。在代谢网络建模的特定情况中,节点代表酶,而边指向相邻的酶,其中一种酶的产物是另一种酶的底物[32]。 大型代谢途径图已经存在数十年[33],虽然还存在相当大的差距需要通过直接实验研究来填补,但是代谢网络可能是所有生物网络中最全面的[2]。研究者已经为很多物种编制了代谢网图,主要是原核生物和单细胞真核生物[34],人类的全面代谢重建也正在进行中[35],还有构建人类特定组织的代谢网络[36],以及先天性代谢缺陷也被绘制到人类代谢网络[37]。代谢网络的构造需要在计算机的帮助下手工完成,要精心选择描述了以纯化或重构酶为特征的一个或多个代谢反应的实验结果的大量出版物,必要时可以从其他物种的同源酶实验描述中汇编被预测反应[7]。通过组装所有实验证明和被预测反应来产生蛋白质组规模的网络图[38]。 代谢网络的应用包括以下几个方面:用于药物发 现[39];用来发现代谢网络中的致病节点[40-41];用来预测特定癌症的治疗miRNA[42];用于辅助识别合成致死剂量(Synthetic dosage lethality,SDL)[43];用于研究人类代谢网络的底物循环[44];用于研究人体生理、病理和药理等[45]。 基因调控网络(Gene Regulatory Networks):在多数基因调控网络中,节点可以是一个转录因子或一个假定的DNA调整元素,有向边代表转录因子到此类调整元素的物理结合[46]。当转录因子结合一个调整DNA元素时,边是进入边;而当调整DNA元素被转录因子结合时,边是出去边。目前有2种方法适用于基因调控网络的大规模绘制,即酵母单杂交(Y1H)方法和染色质免疫沉淀反应(ChIP)方法[47-48]。Y1H方法是“基因中心”的,而ChiP方法是“蛋白质中心”的,它们从转录因子所得并试图捕获所关联的基因区域[49]。这2种方法是互补的,Y1H方法可以发现新的转录因子,而ChiP方法可以发现新的调整主题[50-51]。研究者已经生成了大规模线虫的Y1H网络[52],以及基于ChiP的酵母大规模网络[48],并且已经进行哺乳动物组织细胞培养[53],目前已经建立了针对特定疾病的人类基因调控网络[54-61]。 除了转录因子活动外,整体基因转录水平还通过微RNA(miRNA)进行后转录调整[62]。由miRNA和它们的目标相互作用形成的网络中,节点是一个miRNA或者是目标3端UTR(非转译区),边代表miRNA到目标RNA的互补退火[7]。边是进入的(miRNA结合一个3端UTR元素)或出去的(3端UTR元素被一个miRNA结合)[63]。miRNA的目标一般是可计算预测的。由于转录因子调整miRNA的表达,因此可能把Y1H方法和可计算预测的miRNA/3端UTR相结合,这是一种用来在线虫中获得大规模miRAN框架且可以扩展到其他基因组的策略[63]。 磷酸化和蛋白质在翻译后修饰(Post-Translational Modification,PTM)中是常见的,并且PTM之间的串扰参与了许多疾病。Zhang等[64]构建了一个翻译后调控网络(Post-Translational Regulatory Network,PTRN),其中节点是酶或底物。如果有文献报道了某种酶和其底物,那么建立一个由该酶到其底物的有向连接。在这种方式中,大部分边是从激酶或蛋白酶到其底物蛋白的单向连接。如果一对酶(无论是激酶或蛋白酶)被报告在公共数据库中相互调节,它们之间的边将被表示为一个双向链路。根据PTRN可以确定某些功能性的网络模式,这些模式是药物靶标显著丰富的,其中一些被进一步发现含有多个组合药物靶向的蛋白质。这些发现暗示网络模式可能被用来在设计新药时预测靶标。受此启发,Zhang等[64]提出一个新的计算方法NetTar,用识别出的网络模式来预测药物靶标。实际数据的基准测试结果表明,该方法可以用来精确预测由已知药物靶向的新蛋白质。 基因调控网络主要应用于:识别T细胞相关疾病的早期调节基因[54];构造特定疾病的miRAN靶基因调控网络以识别有效的靶向治疗的靶点[55,64];识别癌症相关基因和模式[56];预测特定疾病的转录因子[57];发现功能模块和枢纽基因[65];分析特定癌症的攻击行为[60];识别特定组织的中心转录因子[66];识别调节基因[67];识别信号蛋白质等[68]。 图2是提供“骨架”信息的3个网络,包括蛋白质相互作用网络、代谢网络和基因调控网络。"
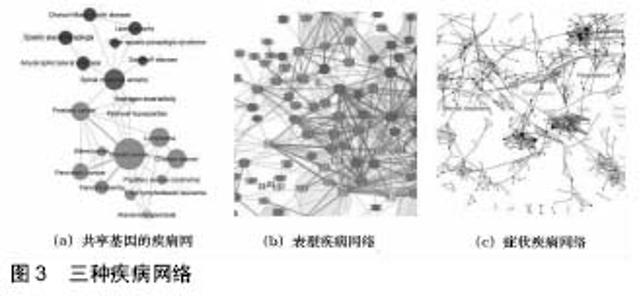
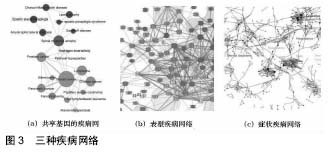
3种生物网络的模块性质:大多数生物网络的节点分布是不均匀的,很多节点密集成团,形成拓扑模块,模块中的节点比该模块外的节点具有更高趋势和相同模块中的节点连接[69]。网络中的潜在拓扑模块可以通过网络聚类算法来确定。这些拓扑模块被认为是执行特殊细胞功能,因此也称为功能模块,即一个在相同网络近邻中的相似或相关功能的节点的聚集[7]。疾病模块表示其扰动导致人类的特定疾病表型的网络成分组[2]。鉴于分子相互作用网络的不完整性和疾病相关基因的知识的限制,可用数据是否具有足够的覆盖以绘制和每个疾病相关的模块还是不明显的。Menche等[70]推导出识别疾病模块的数学条件,并显示每种疾病的基于网络的局部模块决定它和其他疾病的病理关系。例如,具有重叠网络模块的疾病显示显著的共表达模式、症状相似性和合并症,而位于分离的网络邻域的疾病则具有不同表型。 在一个 PPI网络中,不同时间和空间阶段通过相互作用完成某一特定分子进程的蛋白质集合称为蛋白质功能模块。PPI网络功能模块检测的流程通常包括5个步骤: 数据预处理、PPI网络形成、聚类算法、后处理、功能模块输出。根据所采用的计算模型和机制,可以将聚类算法划分为基于传统图理论的方法和基于非传统图理论的方法。基于传统图理论的方法包括基于密度的方法、基于层次的方法和基于划分的方法;而非传统图理论的方法包括基于流模拟的方法、基于谱分析的方法、基于核心-附属关系的方法和基于群集智能的方法[71]。模块检测结果评价方法主要有3种:第2种是信息提取和机器学习常用的评价指标,包括精度、召回率和F度量;第2种是新近提出用来评价模块检测方法性能的度量指标,包括灵敏度、阳性预测率和准确度;第3种是p-值度量,即功能同质性指标[71]。 2.1.2 功能连接的网络 转录谱网络(Transcriptional Profiling Networks):人们期望在共同信令层级或者蛋白质复合体中共同起作用的基因产物在它们的表达模式中比随机的基因产物集合显示更强的相似性[7]。基于很多不同物种的大量多样基因的和环境的条件下的更大纲要的基因表达数据集上构建的共表达网络中,节点代表基因,边连接了那些表现出超过某个设定阈值的相关共表达的基因对[72]。通常使用皮尔森相关系数来衡量任意关联测量,需要使用合适的滴定过程来设定共表达和非共表达之间的阈值[73]。结合共表达谱的物理蛋白质-蛋白质相互作用图,揭示相互作用蛋白质比不相互作用蛋白质更可能是由具有相似表达的基因编码的[72]。这种观察在诸如酵母等很多生物中得到确认[74],但仍然有很多有效的生物相关的蛋白质-蛋白质相互作用对应表达是不相关甚至是反相关的的基因对[7]。 表型谱网络(Phenotypic Profiling Networks):编码功能相关产物的那些基因的扰动经常会赋予相似的表型。基因和表型的关系信息可以看作是矩阵,由一种生物的所有基因和在给定表型谱纲要中被测试的所有表型组成。在获得的表型相似性或“表型”网络中,节点表示基因,边连接那些表现出超出一个设定阈值的相关表型谱的基因对。这里需要滴定来决定什么是表型相似和表型不相似之间的阈值[8]。有助于解释蛋白质-蛋白质相互作用网络的表型谱或“表型”网络的最早证据是从研究线虫DNA破坏反应和两性生殖的研究中获得的[75]。虽然相关表型谱和物理蛋白质-蛋白质交互之间存在强交叠,但是表型网络的物理基础还没有被完整定义[8]。二元蛋白质-蛋白质相互作用、共表达、表型谱3种类型网络的交叠产生了具有高预测能力的整合网络,如线虫早期胚胎形成所表现的那样[8]。 基因相互作用网络(Genetic Interaction Networks):功能相关的基因对趋向于表现出基因相互作用,这是通过比较基因对中的双突变赋予的表型和该双突变的任何一个单突变赋予的表型来定义的[7]。基因相互作用分为正向和负向。负向就是恶化、综合征或致死。当双突变的表型比通过单突变预期的表型显著更糟时就是负向的。正向就是减轻或抑制。当双突变的表型比通过单突变预期的表型显著更好时就是正向的[76]。当基因相互作用不是必须对应相应基因产物的物理相互作用时,它们趋向于揭示参与并行途径或不同分子机制的基因对[76]。负向基因相互作用趋向于既不和蛋白质复合体中的蛋白质关联相关,也不和二元蛋白质-蛋白质相互作用中的蛋白质关联相关;而正向基因相互作用趋向于指向物理上彼此关联的基因产物对[19]。 近十几年来,功能性基因组学的显著进步允许系统化高通量绘制基因相互作用以产生大规模网络[77]。基因相互作用的模式可以用来定义一种类似于表型谱或表型网络这样的网络。类似转录网络和表型谱网络,这种信息可以被看作一种矩阵,它包含一种生物的所有基因和那些呈现出基因相互作用的基因。在这样的“基因相互作用谱”网络中,边功能性地连接两个基于基因相互作用谱的高度相似的基因。当这样的网络和其他类型的相互作用网络结合起来时可以获得生物过程的预测模型[7]。 2.1.3 其他生物分子相互作用网络 Rai等[78]构造了一个获得性高脂血症的脂质-蛋白质-蛋白质相互作用网络(Lipid-Protein-Protein Interaction Network,LPPIN)。胆固醇、甘油二酯、磷脂酰肌醇磷酸双酯和三磷酸肌醇被确定为LPPIN中影响信号通路的核心血脂。丝氨酸/苏氨酸蛋白激酶(Akt)是一个非常重要的中心蛋白。在LPPIN中识别出来的疾病簇是心血管疾病、癌症、阿尔茨海默病和Ⅱ型糖尿病。商业上批准的高血脂症的药物靶标可能潜在地作为特定疾病的治疗目的。Rai等报告了10个潜在的可能介导高血脂症到相应疾病状态的药物靶点。与获得性高脂血症相关的脂质和识别出来的每个疾病簇可能是有用的预兆指纹。这些研究结果提供了一个综合性的脂质-蛋白质相互作用的观点,这种相互作用导致获得高脂血症和相关疾病,并可能证明在未来的转化药学研究上是有用的。 2.2 疾病网络(Diseasome) 人类疾病网络,也称为diseasome,其中节点是疾病,2种疾病通过它们共享的相关细胞成分、生物过程或其他疾病相关因素连接起来。以细胞成分或其他疾病相关因素为节点,边表示某种疾病,如果两个节点和相同疾病相关,那么它们被连接起来,所形成的网络是疾病网络的补充。2种或2种以上疾病可能共享包括基因、蛋白质、miRNA等细胞成分、诸如代谢这样的生物过程及包括表型、症状和环境因素等疾病相关因素。揭示疾病之间的这种连接不仅有助于理解不同医学分支学科称呼的不同表型在分子水平是相连的,而且还有助于理解为什么某些疾病组出现在一起[79]。从疾病网络中挑选出来的共同发病条件为可能产生新的方法来进行疾病预防、诊断和治疗提供了洞察力。基于疾病网络的方法还可以帮助进行药物发现,特别是当碰到使用已批准的药物来治疗分子相连的疾病时[2]。目前人类疾病网络根据其构造基础分为:共享基因、共享代谢、共享miRNA、共享表型、共享症状、共享环境因素等。 2.2.1 共享基因的人类疾病网络 在共享基因的人类疾病网络(Human Disease Network,HDN)中,节点表示疾病,如果2种疾病共享至少一个基因,其突变和这2种疾病相关,那么这2种疾病被连接起来[80]。这个疾病网反映的是疾病共同的基因起源,它是通过从OMIM (Online Mendelian Inheritance in Man)数据库中收集到的基因-疾病联系建立的。在这个疾病网中,1 284种具有相连基因的疾病中的867种至少和一种其他疾病相连,516种属于同一个疾病簇。例如,癌症形成一个紧密的相互连接且容易发现的聚类,通过一小群和多个癌症相联系的基因聚集在一起,例如P53,KRAS,ERBB2或NF1[80]。 和通过共享基因来联系疾病相反,在疾病基因网络(disease gene network,DGN)中,节点代表疾病基因,如果2个基因和相同疾病相关,那么把它们连接起来。该网络反映基因导致的共同疾病[80]。HDN和疾病基因网络这2个网络有助于理解所有已知疾病基因和表型的联系,并在单个框架中进行展示[80]。 2.2.2 代谢疾病网络(Metabolic Disease Network,MDN) 经典的代谢疾病和导致某种代谢酶不表达、不激活或功能受损的突变相关。由于连续的代谢反应在功能上是相互关联的且在一定程度上是同步的,因此这种相互依赖的结果是在某个反应中导致错误的一种酶缺陷可能潜在影响一个或几个后续反应的通量。这样的连锁效应还可能耦合与后续反应相关的代谢疾病,导致合并症效应(意味着不同疾病表型的共表达)。因此,疾病和特定生物化学反应之间的联系还可能导致和同一个途径相关的疾病之间的直接联系[81]。假设给定可能影响耦合反应的代谢缺陷引起的2种疾病,它们的病理表型也可能是相关的。为支持这种假设所建立的代谢疾病网络中,每个节点对应一种疾病,如果和它们相关的代谢反应是邻近的,那么它们被连接,意味着它们的通量是耦合的[81]。代谢疾病网络的可见明显聚类反映不同的代谢路径。例如,嘌呤代谢由62种反应组成,和包括核苷磷酸化酶缺乏症在内的33种疾病相关。这些疾病形成一个可见的清晰聚类。合并症分析证实了代谢耦合的功能相关性:和那些没有代谢相关联系的疾病对相比,在代谢疾病网络中相连的疾病对具有1.8倍增长的合并症。如果各自疾病基因催化的反应流量是自耦合的,那么合并症就更加明显,即一个通量中的改变引起其他通量的显著改变,即使对应的反应是不邻近的[2]。 2.2.3 基于miRNA的疾病网络(miRNA-based disease network) miRNA是一类小的非编码RNA,它的通常功能是作为转录后目标mRNA表达的负向调控器,导致目标mRNA降解或翻译抑制。越来越多的证明表明miRNA在很多关键生物过程中起关键作用,例如细胞生长、组织分化、细胞增殖、胚胎发育和细胞凋亡,因此miRNA的突变、生物合成的功能失灵、调控失灵,会导致其调控的mRNA异常,从而引发各种疾病。Lu等[82]基于一个miRNA列表和来自HMDD(human miRNA-associated disease database)的人类疾病列表之间的连接建立一个二分图;然后基于该二分图构建miRNA相关疾病网络(miRNA-associated disease network,MDN),如果2种疾病共享至少一个公共相关miRNA,则把它们连接起来。代谢疾病网络显示了簇结构,其中相似疾病被聚集在一起。所有癌症被联系在一起,表明各种癌症可能在miRNA水平共享相似联系,其中某些强烈的致癌miRNA或miRNA抑制剂可能起关键作用。例如,各种研究表明miR-21在各种癌症中是过表达的,而miR-125a在各种癌症中显示反向调节,表明它是miRNA的抑制剂。 Yuan等[83]通过整合miRNA靶向相互作用(miRNA- target interaction,MTIs)、蛋白质-蛋白质相互作用、基因-疾病联系等数据,并定量考虑靶向疾病相关基因的miRNA的直接影响和由蛋白质-蛋白质相互作用触发的间接影响以识别潜在功能miRNA疾病联系(miRNA-disease association,MDA),发现了19 648种假定MDA,其中10%是被实验验证的;还发现一个miR-24和卵巢癌之间的新MDA。 2.2.4 表型疾病网络(Phenotypic Disease Network,PDN) 表型疾病网络是通过直接观察疾病之间的合并症来连接疾病对而构造的。Rzhetsk等[84]在哥伦比亚医学中心的150万个疾病历史中推断161个疾病之间的合并症链接。Hidalgo等[85]从3 000万个医疗保险病人的疾病历史中建立了一个包括657个疾病的网络。在这些图中,如果两个疾病的合并症超出预先定义的阈值,那么它们被连接。表型疾病网络不关心所观察到的合并症的机制,这些合并症可能是源于在分子水平的相关性(正如从HDN、代谢疾病网络或基于miRNA疾病网络所观察到的),或是源于网络环境的或治疗相关的扰动。然而表型疾病网络捕获了疾病进程,因为患者往往在他们所患疾病的网络近邻发展疾病。而且,那些被诊断出患有表型疾病网络中具有更多连接的疾病的患者比那些具有更少联系的疾病的患者具有更高的死亡率。Van Driel等[86]建议表型信息的另一种用法是建立疾病网络,他利用文本挖掘为OMIM数据库中超过5 000种人类表型分配一个来自医学学科标题词汇的表型特征字符串。他们的表型描述的重叠被用来连接不同的疾病,发现表型相似性和两个相连疾病的分子特征的正相关,从蛋白质序列水平的相关性到蛋白质模式和与疾病相关的蛋白质之间的直接蛋白质-蛋白质相互作用。 2.2.5 症状疾病网络(Human Symptoms Disease Network,HSDN):疾病的分子来源和它们导致的症状之间的关系对于医学研究来说是一个关键任务。Zhou等[87]使用一个大型生物医学文献数据库PubMed构建一个基于症状的人类疾病网络,用术语频率-逆文档频率来量化症状和疾病之间的关系,获得具有七百多万条 4 219种疾病之间的正相似性连接的症状疾病网络。症状疾病网络覆盖所有MeSH(Medical Subject Heading)疾病目录,从诸如癌症这样的大类到像脑海绵状血管瘤这样的情况。症状疾病网络组成了一个巨大的组成部分,即所有疾病直接或间接和其他所有疾病相连。这个网络是非常密集的,94%的节点和超过50%的其他节点相连。最被高度连接的疾病是低钠血症(4 214种疾病的邻居),电解质紊乱和很多常见症状相连,这些症状包括头痛、恶心和疲劳。具有最少连接的疾病是牙瘤(8种疾病的邻居),一种牙齿引发的肿瘤。Zhou等[87]还研究临床表现和它们底层的分子相互作用之间的联系,发现2种疾病的基于表型相似性和共享基因联系的数量及相关蛋白质相互作用的程度具有强烈相关性;而且一种疾病的临床表型的多样性可以和底层蛋白质相互作用网络的连接模式相关。 2.2.6 基于基因和环境因素的疾病网络(Etiome网络)基因的和环境的因素都可以导致人类疾病。基因的影响对单个疾病是相对好描述的,但缺乏对环境因素扩展列表的整理。Liu等[88]从一个MEDLINE文献的MeSH注释的综合研究中,识别出和3 159种疾病相关的3 342种环境致病因素,以及和来自NIH基因相关数据库(Genetic Association Database,GAD)的1 034种复杂疾病相关的1 100种基因。863种疾病具有基因的和环境的致病因素。整合基因的和环境的因素得到“Etiome”,即疾病病因的综合性纲要。对基因的和环境的致病因素二者进行聚类就是以量化方式把基因放到环境的上下文中。 Etiome网络表明影响同一个器官系统的致病因素趋向于聚集在一起。一个例子是包含1型糖尿病、2型糖尿病和高血糖的聚类。这3种疾病有明显不同的病因,然而它们导致了相同的一组并发症,包括加速动脉粥样硬化、神经病变、视网膜病变、肾病和感染。就是这个共享并发症谱使得这3种疾病集群在彼此旁边。这个例子还说明某些疾病也可能成为其他疾病的致病因素。然而某些关联不是那么清晰。例如,睡眠呼吸暂停、钙化和先天代谢异常也是处在包含糖尿病、高血糖和高血脂的簇中。对该簇的进一步研究显示3种病理过程,但是这3个看起来独立的过程却导致心血管后遗症。这些致病因素导致神经系统后遗症和术后并发症的相似性也有助于集群。这一结果表明睡眠呼吸暂停和糖尿病的患者可能存在高风险的心血管疾病的死亡率和发病率。Liu等[88]还研究了遗传因素在Etiome中的位置,集中在和具有最高文章数的863种疾病相关的5个基因:TP53、ctl4、TNF、ACE和NOS3。研究结果表明这五个基因落入了“Etiome”3个不同的领域:肿瘤、炎症与免疫、代谢。TP53是与其他各种致癌物质聚集在一起的;CTLA4和TNF与免疫介导的过程及甲苯、空气污染等聚集在一起;NOS3和ACE是与炎症、高胰岛素血症和代谢性疾病相关的。 2.2.7 基于差异共表达的疾病网络(The DCE-based Disease Network) 近年来,根据转录行为来估计疾病相似性显著增强了发现新的疾病关系的可能性,而当前可用研究一般通过差异表达分析来挖掘表达数据,差异表达分析已被认为在揭开调控关系失调和疾病的因果方面机会微乎其微[89],因此Yang等[89-90]基于mRNA表达数据和基于方法差异共表达(Differential CoExpression,DCE)分析建立了一个人类疾病网络,包括108种疾病的1 326个有意义的疾病-疾病联系。和通过基于方法的差异表达分析捕获的疾病联系相比,Yang等建立的疾病连接更显著地共享已知疾病基因和药物。他们还发现了一些新的疾病关系,例如肥胖和癌症、肥胖和牛皮癣、肺腺癌和肺炎,这已被普遍认为是彼此无关的,但最近发现有类似的分子机制。此外,发现疾病类型和被影响组织的类型都影响疾病相似性的程度。 图3是3种疾病网络,分别是共享基因的疾病网络、表型疾病网络和症状疾病网络。"
2.3 药物网络层(Drugsome) Diseasom思想的一个自然延伸是把药物包括进行,形成所谓“Drugome”[91]。Yildirim 等[92]使用FDA批准的药物和它们的靶标来产生一个药物-蛋白质相互作用的二分图,如果一种蛋白质是一种药物的靶标,那么它们被连接,由此产生“药 物-靶标网络”(Drug-Target network,药物-靶标网络)。基于药物-靶标网络产生两个生物上相关的网络,即药物网络和靶标-蛋白质网络。在药物网络中,如果2种药物共享至少一个靶标蛋白质,那么它们被相互连接。如果多数药物特定地靶向单一蛋白质,那么药物-靶标网络包含具有很少的或是没有边的孤立节点,但是药物网络却显示不同药物和药物类之间的很多连接。890种批准药物中的788种至少和其他药物有一条连接,即它们和其他药物共享靶标。476种药物被包含在网络的最大相互连接成分中。根据解剖治疗化学(Anatomical Therapeutic Chemical,ATC) )分类对药物-靶标网络中的节点上色。虽然药物-靶标网络的设计独立于任何药物分类知识,所获得的网络自然地和可见地被主要治疗类集群。最明显的例子是大的紧密的神经药物簇。相反,抗肿瘤药物和代谢疾病药物没有形成单个清晰的簇。这些类在大成分中未被充分代表,却在更小的成分中过多代表,体现了相对于它们的类大小的最少连接药物类。 在“靶标-蛋白质网络”(Target-Protein network,PT网络)中,节点代表蛋白质,如果2种蛋白质都被至少一种相同药物靶向,那么它们被连接[92]。靶标-蛋白质网络提供了一种补充,以蛋白质为中心的药理学观点。在靶标-蛋白质网络中,394种中的305种靶标蛋白质和其他靶标蛋白质相连。多靶标药物是造成靶标-蛋白质网络高度相互连接的原因。图4显示了药物-靶标网络和PT网络。"
| [1] Network medicine. From Wikipedia, the free encyclopedia. https://en.wikipedia.org/wiki/Network_medicine[2] Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56-68.[3] Barabási AL. Network medicine-from obesity to the “diseasome”. N Engl J Med. 2007;357(4):404-407.[4] Chan SY, Loscalzo J. The emerging paradigm of network medicine in the study of human disease. Circulation Res. 2012;111(3), 359-374.[5] Sanchez C, Lachaize C, Janody F, et al."Grasping at molecular interactions and genetic networks in Drosophila melanogaster using FlyNets, an Internet database". Nucleic Acids Res. 1999;27(1): 89-94. [6] Interactome.From Wikipedia, the free encyclopedia. https://en.wikipedia.org/wiki/Interactome[7] Vidal M, Cusick ME, Barabási AL.Interactome Networks and Human Disease. Cell. 2011; 144(6): 986-998.[8] Gunsalus KC, Ge H, Schetter AJ, et al.Predictive models of molecular machines involved in Caenorhabditis elegans early embryo genesis. Nature.2005;436, 861-865.[9] Rigaut G, Shevchenko A, Rutz B,et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999; 17:1030-1032.[10] Charbonnier S, Gallego O, Gavin AC. The social network of acell: recent advances in interactome mapping. Biotechnol Annu Rev. 2008;14:1-28.[11] Kuhner S, van Noort V, Betts MJ, et al. Proteome organization in a genome-reduced bacterium. Science.2009;326:1235-1240.[12] Seebacher J, Gavin AC. SnapShot: Protein-protein interaction networks. Cell. Cell. 2011;144(6):1000, [13] RRual JF, Venkatesan K, Hao T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005; 437(7062):1173-1178.[14] Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell.2005;122(6):957-968.[15] Sun J, Zhao Z.A comparative study of cancer proteins in the human protein-protein interaction network.BMC Genomics. 2010 Dec 1;11 Suppl 3:S5.[16] Schaefer MH, Lopes TJ, Mah N, et al. Adding Protein Context to the Human Protein-Protein Interaction Network to Reveal Meaningful Interactions. PLoS Comput Biol. 2013;9(1): e1002860.[17] Rolland T, Ta?an M, Charloteaux B,et al.A Proteome-Scale Map of the Human Interactome Network.Cell. 2014;159(5):1212-1226.[18] Venkatesan K, Rual JF, Vazquez A, et al. An empirical framework for binary interactome mapping. Nat. Methods. 2009;6: 83-90[19] Costanzo M, Baryshnikova A, Bellay J,et al. The genetic landscape of a cell. Science.2010; 327:425-431.[20] Li BQ, Huang T, Liu L,et al. Identification of Colorectal Cancer Related Genes with mRMR and Shortest Path in Protein-Protein Interaction Network. PLoS ONE.2012;7(4): e33393.[21] Paul Y, Hasija Y. Gene Prioritization by Integrated Analysis of Protein Structural and Network Topological Properties for the Protein-Protein Interaction Network of Neurological Disorders. Scientifica (Cairo). 2016;2016:9589404.[22] Shityakov S, Dandekar T, Forster C. Gene expression profiles and protein-protein interaction network analysis in AIDS patients with HIV-associated encephalitis and dementia. HIV AIDS(Auckl). 2015;7:265-276. [23] Safaei A, Rezaei Tavirani M, Arefi Oskouei A,et al. Protein-protein interaction network analysis of cirrhosis liver disease. Gastroenterol Hepatol Bed Bench. 2016;9(2):114-233.[24] Shityakov S, Dandekar T, Förster C.Gene expression profiles and protein–protein interaction network analysis in AIDS patients with HIV-associated encephalitis and dementia.HIV AIDS (Auckl). 2015;7:265-276. [25] vinayagam A,Gibson TE,Yilmazel B,et al.Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets. Proc Natl Acad Sci USA. 2016;113(18):4976-4981. [26] Chapple CE, Robisson B, Spinelli L, et al. Extreme multifunctional proteins identified from a human protein interaction network.Nat Commun. 2015;6:7412. [27] Zhang C,Shen L.Functional modules analysis based on protein-protein network analysis in ankylosing spondylitis.Eur Rev Med Pharmacol Sci. 2012;16(13):1821-1827.[28] Zhao Y, Mooney SD. Functional organization and its implication in evolution of the human protein-protein interaction network.BMC Genomics. 2012;13:150.[29] Schuster S, Fell DA, Dandekar T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000;18:326-332.[30] Edwards JS, Ibarra RU, Palsson BO. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat.Biotechnol. 2001;19;125-130.[31] Jeong H, Mason SP, Barabasi AL,et al. Lethality and centrality in protein networks. Nature.2001;411:41-42.[32] Lee DS, Park J, Kay KA,et al. The implications of human metabolic network topology for disease comorbidity. Proc. Natl. Acad. Sci. USA.2008;105:9880-9885.[33] Kanehisa M, Araki M, Goto S,et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res.2008;36: D480-D484.[34] Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol.2009; 5:320.[35] Duarte NC, Becker SA, Jamshidi N, et al.Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA.2007; 104:1777-1782. [36] Karlstädt A, Fliegner D, Kararigas G, et al. CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst Biol.2012;6:114. [37] Sahoo S,Franzson L,Jonsson JJ,et al. A compendium of inborn errors of metabolism mapped onto the human metabolic network. Mol Biosyst. 2012;8(10):2545-2558.[38] Mo ML,Palsson BO.Understanding human metabolic physiology: a genome-to-systems approach. Trends Biotechnol. 2009;27:37-44.[39] Kell DB, Goodacre R. Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery. Drug Discov Today.2014;19(2):171-182. [40] Galhardo M,Sinkkonen L,Berninger P,et al.Integrated analysis of transcript-level regulation of metabolism reveals disease-relevant nodes of the human metabolic network. Nucleic Acids Res.2014;42(3):1474-1496.[41] Hu J,Locasale JW,Bielas JH,et al.Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol.2013;31(6):522-529[42] Wu M, Chan C.Prediction of therapeutic microRNA based on the human metabolic network.Bioinformatics. 2014;30(8): 1163-1171.[43] Megchelenbrink W, Katzir R, Lu X.Synthetic dosage lethality in the human metabolic network is highly predictive of tumor growth and cancer patient survival.Proc Natl Acad Sci USA. 2015;112(39):12217-12222.[44] Gebauer J, Schuster S, de Figueiredo LF, et al. Detecting and investigating substrate cycles in a genome-scale human metabolic network.FEBS J.2012;279(17):3192-3202[45] Bordbar A, Palsson BO.Using the reconstructed genome-scale human metabolic network to study physiology and pathology. J Intern Med. 2012;271(2):131-141.[46] Zhu C,Byers KJ,McCord RP,et al.High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res.2009;19:556–566.[47] Deplancke B, Dupuy D, Vidal M, et al. A Gatewaycompatible yeast one-hybrid system. Genome Res. 2004;14, 2093–2101.[48] Lee TI, Rinaldi NJ, Robert F,et al. Transcriptional regulatory networks inSaccharomyces cerevisiae. Science.2002;298, 799-804.[49] Walhout AJ. Unraveling transcription regulatory networks by proteinDNA and protein-protein interaction mapping. Genome Res. 2006;16:1445-1454.[50] Reece-Hoyes JS, Deplancke B, Shingles J,et al. A compendium ofCaenorhabditis elegansregulatory transcription factors: a resource for mapping transcription regulatory networks. Genome Biol. 2005;6:R110.[51] Vaquerizas JM, Kummerfeld SK, Teichmann SA, et al. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 2009; 10:252-263.[52] Grove CA, De Masi F, Barrasa MI, et al. A multiparameter network reveals extensive divergence betweenC. elegansbHLH transcription factors. Cell.2009;138: 314-327.[53] Cawley S, Bekiranov S, Ng HH, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell.2004;116:499-509.[54] Gustafsson M, Gawel DR, Alfredsson L,et al.A validated gene regulatory network and GWAS identifies early regulators of T cell-associated diseases. Sci Transl Med. 2015;7(313):313ra178.[55] He C, Gao H, Fan X, et al. Identification of a novel miRNA-target gene regulatory network in osteosarcoma by integrating transcriptome analysis. Int J Clin Exp Pathol. 2015; 8(7):8348-8357.[56] Carson MB, Gu J, Yu G, et al. Identification of cancer-related genes and motifs in the human gene regulatory network. IET Syst Biol. 2015;9(4):128-134.[57] Mohammadnia A, Yaqubi M, Fallahi H. Predicting transcription factors in human alcoholic hepatitis from gene regulatory network. Eur Rev Med Pharmacol Sci. 2015;19(12):2246-2253.[58] Kousa YA, Schutte BC.Toward an orofacial gene regulatory network. Dev Dyn. 2016;245(3):220-232.[59] Tang WW, Dietmann S, Irie N,et al. A Unique Gene Regulatory Network Resets the Human Germline Epigenome for Development. Cell. 2015;161(6):1453-1467.[60] Yang X, Jia M, Li Z, et al. Bioinformatics analysis of aggressive behavior of breast cancer via an integrated gene regulatory network.J Cancer Res Ther.2014;10(4):1013-1018.[61] Xu X, Li H.Integrated microRNA gene analysis of coronary artery disease based on miRNA and gene expression profiles.Mol Med Rep.2016;13(4):3063-3073.[62] Ruvkun G, Wightman B,Ha I. The 20 years it took to recognize the importance of tiny RNAs. Cell.2004;116;S93-S96.[63] Martinez NJ, Ow MC, Barrasa MI,et al. AC. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008;22: 2535-2549.[64] Zhang XD, Song J, Bork P, et al.The exploration of network motifs as potential drug targets from post-translational regulatory networks.Scientific Reports.2016;6:20558.[65] Zamani-Ahmadmahmudi M, Najafi A, Nassiri SM, et al. Reconstruction of canine diffuse large B-cell lymphoma gene regulatory network: detection of functional modules and hub genes. J Comp Pathol. 2015;152(2-3):119-130.[66] Gubelmann C, Schwalie PC, Raghav SK, et al. Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. Elife. 2014;3:e03346.[67] Janky R, Verfaillie A, Imrichová H, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014;10(7):e1003731.[68] Dusonchet J, Li H, Guillily M, et al. A Parkinson's disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum Mol Genet.2014;23(18):4887-4905.[69] Ravasz E,Somera AL,Mongru DA,et al. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551-1555.[70] Menche J, Sharma A, Kitsak M, et al.Uncovering Disease-disease Relationships Through the Incomplete Interactome. Science.2015;347, 1257601.[71] 冀俊忠,刘志军,刘红欣,等. 蛋白质相互作用网络功能模块检测的研究综述[J].自动化学报, 2014, 40(4):577-593.[72] Kemmeren P, van Berkum NL, Vilo J, et al. Protein interaction verification and functional annotation by integrated analysis of genome-scale data. Mol. Cell.2002; 9:1133-1143.[73] Stuart JM, Segal E, Koller D,et al. A gene-coexpression network for global discovery of conserved genetic modules. Science.2003;302:249-255.[74] Ge H, Walhout AJ, Vidal M. Integrating ‘omic’ information: a bridge between genomics and systems biology. Trends Genet. 2003;19: 551-560.[75] Boulton SJ, Gartner A, Reboul J, et al. Combined functional genomic maps of theC. elegansDNA damage response. Science. 2002;295:127-131.[76] Mani R, St Onge RP, Hartman JL,et al. Defining genetic interaction. Proc. Natl. Acad. Sci. USA.2008;105: 3461-3466.[77] Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat. Rev. Genet. 2007; 8:437-449.[78] Rai S, Bhatnagar S. Hyperlipidemia, Disease Associations, and Top 10 Potential Drug Targets: A Network View. OMICS. 2016; 20(3):152-168.[79] Network medicine. From Wikipedia, the free encyclopedia. https://en.wikipedia.org/wiki/Network_medicine#Diseasome.[80] Goh KI, Cusick ME, Valle D, et al. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685-8690.[81] Lee DS, Park J, Kay KA, et al. The implications of human metabolic network topology for disease comorbidity. PNAS. 2008; 105:9880-9885.[82] Lu M, Zhang Q, Deng M, et al. An Analysis of Human MicroRNA and Disease Associations. PLoS One. 2008;3(10): e3420. [83] Yuan D, Cui X, Wang Y, et al. Enrichment Analysis Identifies Functional MicroRNA-Disease Associations in Humans. PLoS One.2015;10(8): e0136285. [84] Rzhetsky A, et al. Probing genetic overlap among complex human phenotypes. PNAS. 2007;104:11694-11699.[85] Hidalgo C, Blumm N, Barabási AL, et al.A Dynamic Network Approach for the Study of Human Phenotypes. Plos Computational Biology. 2009; 5 e1000353.[86] van Driel MA, Bruggeman J, Vriend G, et al. A text-mining analysis of the human phenome. Eur J Hum Genet. 2006; 14(5):535-542.[87] Zhou X, Menche J, Barabási AL, et al.Human symptoms-disease network.Nat. Commun. 2014;5:4212.[88] Liu YI, Wise PH, Butte AJ. The "etiome": identification and clustering of human disease etiological factors. BMC Bioinformatics. 2009; 10:S14.[89] Yang J, Wu SJ, Dai WT, et al. The human disease network in terms of dysfunctional regulatory mechanisms. Biol Direct. 2015;10:60.[90] Yang J, Wu SJ, Yang SY, et al.DNetDB: The human disease network database based on dysfunctional regulation mechanism. BMC Syst Biol. 2016;10(1):36.[91] Goh KI,Choi IG. Exploring the human diseasome: the human disease network. Brief Funct Genomics. 2012;11(6):533-542.[92] Yildirim MA, Goh KI, Cusick ME, et al. Drug-target network. Nat Biotechnol.2007; 25:1119-1126. |
| [1] | Yao Xiaoling, Peng Jiancheng, Xu Yuerong, Yang Zhidong, Zhang Shuncong. Variable-angle zero-notch anterior interbody fusion system in the treatment of cervical spondylotic myelopathy: 30-month follow-up [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(9): 1377-1382. |
| [2] | An Weizheng, He Xiao, Ren Shuai, Liu Jianyu. Potential of muscle-derived stem cells in peripheral nerve regeneration [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1130-1136. |
| [3] | Zhang Jinglin, Leng Min, Zhu Boheng, Wang Hong. Mechanism and application of stem cell-derived exosomes in promoting diabetic wound healing [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1113-1118. |
| [4] | He Yunying, Li Lingjie, Zhang Shuqi, Li Yuzhou, Yang Sheng, Ji Ping. Method of constructing cell spheroids based on agarose and polyacrylic molds [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 553-559. |
| [5] | He Guanyu, Xu Baoshan, Du Lilong, Zhang Tongxing, Huo Zhenxin, Shen Li. Biomimetic orientated microchannel annulus fibrosus scaffold constructed by silk fibroin [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 560-566. |
| [6] | Chen Xiaoxu, Luo Yaxin, Bi Haoran, Yang Kun. Preparation and application of acellular scaffold in tissue engineering and regenerative medicine [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 591-596. |
| [7] | Kang Kunlong, Wang Xintao. Research hotspot of biological scaffold materials promoting osteogenic differentiation of bone marrow mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 597-603. |
| [8] | Shen Jiahua, Fu Yong. Application of graphene-based nanomaterials in stem cells [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 604-609. |
| [9] | Zhang Tong, Cai Jinchi, Yuan Zhifa, Zhao Haiyan, Han Xingwen, Wang Wenji. Hyaluronic acid-based composite hydrogel in cartilage injury caused by osteoarthritis: application and mechanism [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 617-625. |
| [10] | Li Hui, Chen Lianglong. Application and characteristics of bone graft materials in the treatment of spinal tuberculosis [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 626-630. |
| [11] | Gao Cangjian, Yang Zhen, Liu Shuyun, Li Hao, Fu Liwei, Zhao Tianyuan, Chen Wei, Liao Zhiyao, Li Pinxue, Sui Xiang, Guo Quanyi. Electrospinning for rotator cuff repair [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(4): 637-642. |
| [12] | Guan Jian, Jia Yanfei, Zhang Baoxin , Zhao Guozhong. Application of 4D bioprinting in tissue engineering [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(3): 446-455. |
| [13] | Liu Jiali, Suo Hairui, Yang Han, Wang Ling, Xu Mingen. Influence of lay-down angles on mechanical properties of three-dimensional printed polycaprolactone scaffolds [J]. Chinese Journal of Tissue Engineering Research, 2022, 10(16): 2612-2617. |
| [14] | Huang Bo, Chen Mingxue, Peng Liqing, Luo Xujiang, Li Huo, Wang Hao, Tian Qinyu, Lu Xiaobo, Liu Shuyun, Guo Quanyi . Fabrication and biocompatibility of injectable gelatin-methacryloyl/cartilage-derived matrix particles composite hydrogel scaffold [J]. Chinese Journal of Tissue Engineering Research, 2022, 10(16): 2600-2606. |
| [15] | Fang Xiaoyang, Tang Tian, Wang Nan, Qian Yuzhang, Xie Lin. Repair and regenerative therapies of the annulus fibrosus [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(10): 1582-1587. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||