Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (28): 7378-7387.doi: 10.12307/2026.823
Previous Articles Next Articles
Metabolic dysfunction-related fatty liver disease: pathological mechanisms mediated by common and heterogeneous pathways
Sun Zhiyuan1, Xu Kai2, Tian Xuewen1, Shang Qinghui1
- 1Shandong Scientific Fitness Guidance Center, Shandong Sport University, Jinan 250102, Shandong Province, China; 2College of Education and Physical Education, Yangtze University, Jingzhou 434000, Hubei Province, China
-
Received:2025-10-15Revised:2025-12-16Online:2026-10-08Published:2026-02-24 -
Contact:Tian Xuewen, PhD, Professor, Shandong Scientific Fitness Guidance Center, Shandong Sport University, Jinan 250102, Shandong Province, China Corresponding author: Shang Qinghui, PhD, Lecturer, Shandong Scientific Fitness Guidance Center, Shandong Sport University, Jinan 250102, Shandong Province, China -
About author:Sun Zhiyuan, PhD, Lecturer, Shandong Scientific Fitness Guidance Center, Shandong Sport University, Jinan 250102, Shandong Province, China -
Supported by:National Science and Technology Major Project - Major Project of Four Major Chronic Diseases, No. 2024ZD0531803 (to TXW)
CLC Number:
Cite this article
Sun Zhiyuan, Xu Kai, Tian Xuewen, Shang Qinghui. Metabolic dysfunction-related fatty liver disease: pathological mechanisms mediated by common and heterogeneous pathways[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(28): 7378-7387.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks

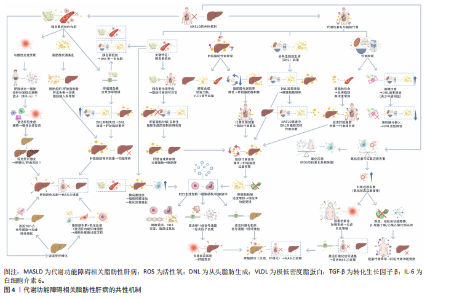
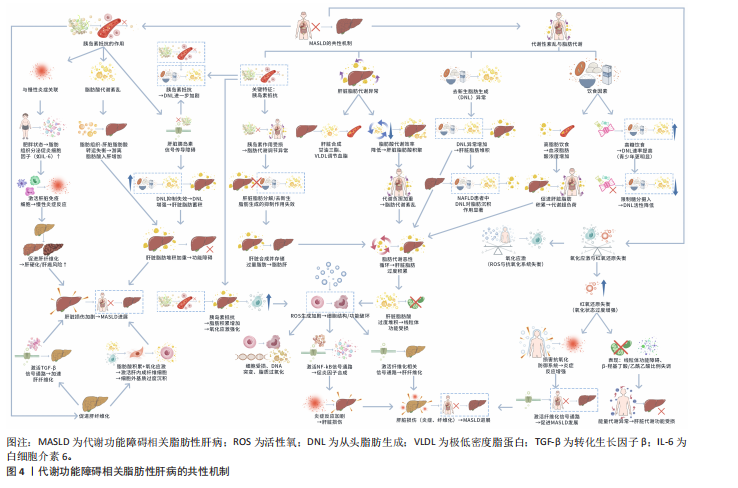
2.1 代谢功能障碍相关脂肪性肝病的病理机制的时间脉络 见表1。 2.2 代谢功能障碍相关脂肪性肝病概述 代谢功能障碍相关脂肪性肝病是一类与代谢紊乱紧密相关的肝脏疾病,特征表现为肝脏脂肪堆积[1,10-11]。代谢功能障碍相关脂肪性肝病表现为脂肪肝,病理特征为肝细胞内脂肪过度沉积,并且常伴随胰岛素抵抗、高血糖、脂质代谢紊乱以及肥胖、2型糖尿病等其他代谢性疾病[2-3]。尽管代谢功能障碍相关脂肪性肝病通常被认为是非酒精性脂肪性肝病的一部分,但独特的代谢紊乱背景和进展机制使该病成为一个独立且愈发重要的临床课题。 代谢功能障碍相关脂肪性肝病的病理过程与个体代谢稳态失衡密切相关,其中胰岛素抵抗和脂质代谢失调构成了该病的病理生理学基础[12]。代谢障碍相关脂肪肝疾病的特点是肝细胞中脂质沉积异常增加,导致肝纤维化、晚期肝硬化和肝细胞癌,该疾病的本质在于肝脏前化学中甘油三酯和其他中性脂质的病理积累,直接导致肝细胞的结构和功能损害;随着疾病的进展,肝脏微环境由慢性炎症反应和氧化应激活[13],协同作用加剧肝结构的病理损害,进而促进肝纤维化[4,14]。 此外,代谢功能障碍相关脂肪性肝病的发病机制复杂并具有异质性,不同个体的临床表现和进展情况可能会有明显差异,部分患者早期只表现为肝脏脂肪堆积,但可能不会出现明显的临床症状或其他代谢并发症[10],然而随着疾病进展,肝脏损伤可能促使炎症、纤维化,甚至肝硬化和肝癌的发生[5]。此外,代谢功能障碍相关脂肪性肝病还可能增加心血管代谢疾病和癌症的风险[10]。 因此,掌握代谢功能障碍相关脂肪性肝病发病机制和与其他相关代谢性疾病的联系,对于早期进行诊断、治疗具有很大意义。目前的研究重点除对代谢功能障碍相关脂肪性肝病基本病理机制的深刻理解外,还需探究干预代谢路径来有效控制疾病的相关进展,预防疾病不良影响以及并发症的产生。 2.3 代谢功能障碍相关脂肪性肝病的共性机制 代谢功能障碍相关脂肪性肝病的共性机制是一个多环节相互作用的复杂网络,核心围绕代谢紊乱、氧化应激及胰岛素抵抗等关键过程展开,这些过程相互交织,共同推动着肝细胞脂肪沉积及后续的病理损伤。其中代谢性紊乱,尤其是脂肪代谢异常,作为共性机制中的基础环节,在代谢功能障碍相关脂肪性肝病的发生发展中发挥着至关重要的作用。代谢功能障碍相关脂肪性肝病的共性机制见图4。 2.3.1 代谢性紊乱与脂肪代谢 代谢性紊乱,尤其是脂肪代谢异常,是推动代谢功能障碍相关脂肪性肝病发展的重要因素。通常来说,代谢功能障碍相关脂肪性肝病患者的主要代谢性特征往往包含有胰岛素抵抗[15],胰岛素抵抗通过损害体内胰岛素作用来影响脂肪代谢的调节[16]。 肝脏作为脂肪代谢的关键器官,能够合成甘油三酯和极低密度脂蛋白,以此来调节血脂水平。胰岛素促进葡萄糖进入细胞供能,并将多余葡萄糖转化为甘油三酯储存于脂肪组织[17],肝脏对胰岛素敏感时能正常分解或储存脂肪;而当胰岛素抵抗时,肝脏对脂肪的处理能力下降,导致脂肪在肝细胞内异常堆积,引发脂肪肝。研究发现,肥胖状态下肝脏会通过氨基酸代谢途径促使新的脂肪生成,进一步影响脂肪存储与代谢平衡[17]。除此之外,研究发"

现,含WD40重复序列的蛋白6在胰岛素抵抗期间促进肝脏从头脂肪生成[18]。 在缺乏外源脂肪酸时,肝脏将碳水化合物合成脂肪的过程称为去新生脂肪生成。在代谢功能障碍相关脂肪性肝病中,导致肝脏脂肪堆积的一个重要因素就是去新生脂肪生成的异常增加[17]。去新生脂肪生成对非酒精性脂肪肝病患者肝脏脂肪沉积的作用效果明显,胰岛素抵抗的产生会加重脂肪性肝病。SMITH等[3]发现,传统非酒精性脂肪性肝病患者脂肪肝的形成与去新生脂肪生成相关联。COHEN等[19]研究发现,高糖饮食可提高肝脏的去新生脂肪生成速率,这种情况在青少年脂肪肝患者中更为明显;而通过限制糖分摄入可显著降低肝脏去新生脂肪生成速率。总体而言,以上研究表明,高糖饮食刺激、新生脂肪生成激活及胰岛素抵抗均能影响并加剧代谢紊乱进程,进而促使脂肪性肝病的发生与发展。 高脂肪饮食通过增加血液中的脂肪酸浓度促进肝脏脂肪的积累,促进非酒精性脂肪肝的形成[20]。此外,脂肪酸的过量供应会使肝脏的代谢能力出现超负荷,导致脂肪代谢紊乱,进一步促进脂肪肝的形成[21]。在代谢功能障碍相关脂肪性肝病患者中,肝脏的脂肪酸代谢效率会降低,这会使脂肪酸在肝脏内进行性积聚,加重肝脏的代谢负担。同时,肝脏的脂肪酸代谢紊乱不仅涉及脂肪积累,还与去新脂合成等代谢途径的异常相关。Nú?EZ-SáNCHEZ等[5]通过病例-对照研究发现,代谢功能障碍相关脂肪性肝病患者的肝脏脂肪酸谱显著改变,而产生这种变化的主要原因为肝脏脂肪生成的增强。并且不当饮食习惯会通过增强肝脏去新脂合成等途径促使代谢功能障碍相关脂肪性肝病的发生以及加剧腹部肥胖,促使肝脏脂肪酸的积累,对代谢功能造成影响[22]。 综上所述,代谢功能障碍相关脂肪性肝病中的代谢性紊乱导致肝脏脂肪代谢恶性循环的形成,在此过程中,除了过量脂肪的积累外,还伴随慢性炎症和氧化应激的作用,导致肝脏损伤。由此可见,改善代谢紊乱与纠正脂肪代谢异常,或许是预防和治疗代谢功能障碍相关脂肪性肝病的关键环节。 2.3.2 氧化应激与氧化还原失衡 肝脏是人体核心代谢枢纽,通过处理脂肪代谢产物、解毒、合成和分解多种生物分子维持稳态[23]。肝脏代谢紊乱在代谢功能障碍相关脂肪性肝病患者中经常伴随氧化应激的增加,该情况与肝脏脂肪积累、肝细胞损伤及疾病密切关联[24]。 氧化应激是指体内自由基(尤其是活性氧种)与抗氧化系统之间的不平衡,导致细胞损伤、DNA突变和脂质过氧化等不良反应[25]。活性氧过多积累会破坏细胞结构和功能,尤其是在代谢功能障碍相关脂肪性肝病患者的肝脏中氧化应激反应更为明显,主要存在于肝脏脂肪沉积的区域[26]。在该情况下,过度堆积的脂肪酸会使线粒体功能受到损害,进一步加剧活性氧的生成,这不仅能够直接损伤肝细胞,还会通过诱导炎症反应和细胞凋亡加剧肝脏损伤[27]。 有研究表明,活性氧与抗氧化系统的不平衡是导致细胞损伤、DNA突变和脂质过氧化等反应的主要原因[27],而在代谢功能障碍相关脂肪性肝病患者中,有关研究也充分证实了肝脏脂肪沉积区域的氧化应激增加[26]。研究证实,脂肪酸氧化产生的活性氧在代谢功能障碍相关脂肪性肝病的进展中扮演着关键角色,其会增强氧化应激反应,进一步影响肝脏功能和健康[26]。尤其是处于去新生脂肪生成的过度活跃状态下,肝脏内的脂肪酸合成增加,之后的氧化应激反应也会随之加剧,该过程可能会导致加重肝脏损伤,甚至促使肝脏纤维化的发生[28]。 在代谢功能障碍相关脂肪性肝病中,氧化还原失衡驱动了显著的氧化应激,进而损害线粒体功能,功能受损的线粒体无法有效进行脂肪酸氧化,从而引发肝内脂质代谢异常,最终导致全身性能量代谢紊乱。这一系列连锁反应在生化层面体现为β-羟基丁酸与乙酰乙酸的比例失调,该指标也因此成为评估线粒体功能与能量代谢状态的重要依据[29-31]。 氧化还原失衡不仅是肝脏脂代谢紊乱的元凶,更是推动代谢功能障碍相关脂肪性肝病炎症与纤维化进程的关键放大器:氧化还原失衡通过激活核因子κB等通路触发并维持一种慢性炎症状态[32-34],这种持续的氧化应激环境不仅直接损伤肝细胞,还会削弱肝脏的抗氧化能力,并激活与纤维化相关的信号通路,从而直接促进肝纤维化的形成[26]。因此,氧化应激既是炎症的催化剂,也是纤维化的启动器,这使得它成为代谢功能障碍相关脂肪性肝病向更严重阶段演变的一个核心枢纽,在急、慢性肝损伤中均起着至关重要的作用[35-36]。 综上所述,氧化应激与氧化还原失衡作为代谢功能障碍相关脂肪性肝病的病理机制和核心构成,直接参与并影响疾病发生与发展的整个过程,因此,调节氧化应激和恢复氧化还原平衡可能是干预和治疗代谢功能障碍相关脂肪性肝病的一个潜在靶点。后续研究指向中应思考如何通过抗氧化治疗或厘正氧化还原失衡,缓解代谢功能障碍相关脂肪性肝病患者肝脏的相关病理进程。 2.3.3 胰岛素抵抗的作用 胰岛素抵抗是代谢功能障碍相关脂肪性肝病发生与进展的核心驱动力[37],其通过破坏肝脏脂质代谢稳态、引发慢性炎症及直接促进纤维化等多重机制共同推动代谢功能障碍相关脂肪性肝病的进程[38]。 正常状态下,胰岛素通过抑制肝脏去新生脂肪生成过程来调节肝脏脂肪的合成[39];而在"

胰岛素抵抗下,肝脏细胞对胰岛素的敏感程度减弱,致使去新生脂肪生成过程得到增强,过量的脂肪酸在肝内合成并蓄积,此过程与脂肪组织脂解增加相叠加,致使大量游离脂肪酸涌入肝脏。在脂肪酸涌入与内源性合成增强的双重压力下,肝细胞脂质代谢超载,甘油三酯异常蓄积,从而形成脂肪肝[40-41]。研究进一步揭示,在肥胖型2型糖尿病患者中,肝脏血小板衍生生长因子AA等信号通路的上调与胰岛素抵抗的发生密切相关,为干预提供了潜在新靶点[16,18]。 胰岛素抵抗与慢性低度炎症状态密切相关,加速代谢功能障碍相关脂肪性肝病的进展。研究发现,肥胖状态下脂肪组织分泌的促炎细胞因子和白细胞介素6增加紧密相关,这些因子可激活肝脏免疫细胞,诱导肝脏的慢性炎症反应[42],同时还会促进肝纤维化进程,提高肝硬化甚至肝细胞癌的发病风险[43]。胰岛素抵抗主要通过以下途径促进肝纤维化的发展:一方面,胰岛素抵抗通过脂肪酸积累与氧化应激强化激活肝内成纤维细胞,致使胶原蛋白等细胞外基质的过度沉积,肝星状细胞因脂肪酸的积累和氧化应激被激活,进而促进肝纤维化进展[44];另一方面,胰岛素抵抗还可通过激活转化生长因子β等相关信号通路进一步加速肝脏纤维化进程[45]。 综上所述,胰岛素抵抗在代谢功能障碍相关脂肪性肝病的发展过程中起关键作用,其通过驱动肝脂肪变性、激活慢性炎症及直接促进纤维化构建了一个推动疾病不断恶化的恶性循环。基于此,改善胰岛素敏感性和控制胰岛素抵抗是治疗代谢功能障碍相关脂肪性肝病的重要方案之一。未来研究方向应聚焦如何通过药物干预或生活方式改变有效改善胰岛素抵抗,从而有效延缓或逆转代谢功能障碍相关脂肪性肝病的进程。 2.4 代谢功能障碍相关脂肪性肝病中不同途径介导的机制 代谢功能障碍相关脂肪性肝病的病理进程并非由单一机制主导,而是通过多种相互关联的途径共同介导,这些途径涵盖遗传调控、代谢通路异常、脂肪组织功能失衡、临床表型差异及遗传与代谢的交互作用等多个维度。在这些复杂的介导途径中,肝脏遗传因素作为独立于代谢紊乱的关键调控力量,通过影响肝脏脂质代谢、线粒体功能等核心过程在代谢功能障碍相关脂肪性肝病的发生发展中占据重要地位。代谢功能障碍相关脂肪性肝病中不同途径介导的机制见表2。 2.4.1 代谢功能障碍相关脂肪性肝病中肝脏遗传介导的机制 肝脏遗传背景是塑造代谢功能障碍相关脂肪性肝病异质性的关键因素,其可显著提升肝脂肪变性、炎症、纤维化及肝细胞癌的风险[46]。例如,携带Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸等位基因的纯合子个体表现出显著的肝脏线粒体功能障碍[47],该障碍会导致去新生脂肪生成减少且碳源代谢向酮体生成方向转变,进而改变肝脏代谢途径[6],这一结论证明遗传变异在肝脏代谢紊乱中起到了关键作用。然而,尽管这些遗传变异对代谢功能障碍相关脂肪性肝病的发病机制有显著影响,但其等位基因携带患者并没有表现出心血管疾病的显著风险增加[48],这一结果说明代谢功能障碍相关脂肪性肝病的发生与心血管疾病之间是间接关联的,并非因果关系。 在一项大规模外显子组相关研究中,多种代谢功能障碍相关脂肪性肝病风险等位基因与肝脏脂肪变性、脂肪性肝炎、肝硬化及肝细胞癌的发展相关联,这些遗传变异不只与代谢功能障碍相关脂肪性肝病的发病风险紧密相关,还与2型糖尿病的发生率增加存在联系,但它们却对低血甘油三酯和低低密度脂蛋白胆固醇浓度具有保护作用,并与心冠动脉疾病风险呈负相关性[49]。尽管已有研究尝试解释其对心血管疾病和胰岛素抵抗的保护作用,但它们与2型糖尿病风险增加的机制仍尚未明确[50]。目前已有研究确认,Patatin样磷脂酶域蛋白3作为一种甘油三酯脂肪酶能够动员多不饱和脂肪酸,以此促进肝脏分泌大颗粒的极低密度脂蛋白[7]。然而,携带Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸等位基因个体,其肝脏中该酶的活性较低,这可能导致肝脏甘油三酯分泌减少,从而导致肝脂肪变性[51];此外,具有Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸等位基因的患者,其肝脏脂质组学特征表现与那些具有肝脂肪变性和胰岛素抵抗的患者明显不同,表现为较少代谢性有害脂质(如饱和脂肪酸和神经酰胺)的积累[52],这表明"


Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸等位基因通过调节肝脏脂肪酸代谢在代谢功能障碍相关脂肪性肝病的进程中发挥着关键作用。基于上述基因中的风险等位基因数量所构建的遗传风险评分,与肝脂肪变性、小叶炎症和纤维化的发生率增加以及去新生脂肪生成降低具有相关性。值得注意的是,尽管这些遗传变异与肝脏代谢紊乱紧密相关,但它们与全身性和脂肪组织的胰岛素抵抗并未表现出直接联系[53]。这一发现证实了代谢功能障碍相关脂肪性肝病的发生不仅仅与遗传因素有关,也与代谢紊乱存在联系,并且两者之间的相互作用关系还需进一步深入讨论。 总体而言,尽管研究揭示了遗传因素在代谢功能障碍相关脂肪性肝病代谢紊乱中的重要作用,但仍有许多问题未得到解释,如代谢功能障碍相关脂肪性肝病与心血管疾病的关系、遗传变异对胰岛素抵抗和糖尿病风险的具体机制等。因此,未来应着重于揭示遗传因素、代谢紊乱与肝脏功能之间的复杂联系,并深入探讨可能的干预策略,以减缓代谢功能障碍相关脂肪性肝病及其相关并发症的进程发展。 2.4.2 代谢功能障碍相关脂肪性肝病中肝脏去新生脂肪生成介导的机制 在代谢功能障碍相关脂肪性肝病的代谢紊乱中,肝脏去新生脂肪生成通路的异常激活已成为一个核心特征,该通路的活性增强在伴有胰岛素抵抗、高血糖和高胰岛素血症的患者中尤为突出,构成了推动疾病发展的重要病理基础[16,54]。DONNELLY等[55]研究发现,代谢功能障碍相关脂肪性肝病患者中,约26%的肝脏甘油三酯来自于去新生脂肪生成,这一关键数据不仅证实了新生脂肪生成在代谢功能障碍相关脂肪性肝病中的作用,也为后续机制的深入探索提供了实证基础。 此外,去新生脂肪生成在代谢功能障碍相关脂肪性肝病中的作用不仅限于贡献脂肪酸的绝对数量,其活性高低更与疾病严重度密切相关。LAMBERT等[56]的研究为此提供了直接证据,该研究证明了肝脏脂肪含量较高患者去新生脂肪酸合成速率是肝脏脂肪含量较低患者的3倍以上,这清晰地揭示了去新生脂肪生成速率与肝脏总脂肪负荷之间存在显著的正相关关系。更重要的是,去新生脂肪生成作为一种受营养调控的代谢通路能被简单碳水化合物高效激活[56],这证明了高糖饮食可通过刺激去新生脂肪生成,进而促进代谢功能障碍相关脂肪性肝病病理的进展。 去新生脂肪生成在代谢功能障碍相关脂肪性肝病中与胰岛素抵抗存在密切的交互关系。SMITH等[3]的研究清晰地揭示了这一关联性,在正常体质量人群中,去新生脂肪生成对肝内甘油三酯棕榈酸的贡献比例为11%,在肥胖人群中这一比例上升至19%,而在肥胖合并代谢功能障碍相关脂肪性肝病患者中,去新生脂肪生成的贡献比例显著升高至38%,这一递增趋势充分表明,胰岛素抵抗的严重程度与去新生脂肪生成的活跃度密切相关,并在代谢功能障碍相关脂肪性肝病的发病中起着作用。此外,去新生脂肪生成在肝脏甘油三酯的生成中具有关键作用,并且在疾病进展至肝纤维化阶段时其贡献率仍保持稳定[57]。这表明在促进肝内脂肪蓄积的同时,肝脏去新生脂肪生成水平与胰岛素敏感性呈显著负相关,而与血浆葡萄糖和胰岛素浓度呈现正相关。因此,脂肪酸的积累并没有削弱去新生脂肪生成与代谢紊乱之间的关联,二者共同构成一个相互促进的代谢轴,可能通过影响胰岛素反应在代谢功能障碍相关脂肪性肝病的疾病进展中发挥重要影响。值得注意的是,去新生脂肪生成的病理意义不仅限于肝脏本身。一项纳入37 358名参与者数据的肝脏甘油三酯输出受损相关遗传变异表明,肝脏脂肪增加的等位基因与冠状动脉疾病和心肌梗死的风险降低相关,但与2型糖尿病的风险升高相关,而与新生脂肪生成增强相关的变异表明,肝脏脂肪增加的等位基因与心肌梗死和冠状动脉疾病的风险升高相关[58]。这表明除了直接参与肝内脂肪代谢外,去新生脂肪生成还可能通过系统性的代谢紊乱成为连接代谢功能障碍相关脂肪性肝病与心血管疾病风险的一个重要桥梁,是导致代谢功能障碍相关脂肪性肝病患者心血管健康风险升高的主要因素之一。因此,深入探索去新生脂肪生成在内的肝脏脂肪代谢途径,不仅能够深化代谢功能障碍相关脂肪性肝病发病机制的理解,也能够为预防和治疗心血管疾病提供新的研究视角和潜在干预靶点。 综上所述,肝脏去新生脂肪生成是代谢功能障碍相关脂肪性肝病的关键代谢途径之一,其活性受胰岛素抵抗与糖代谢异常的直接驱动。现有研究虽确立了去新生脂肪生成的重要性,但其如何与全身性代谢紊乱(如心血管疾病)相互作用,并在疾病不同阶段动态演变的规律,仍未被完全阐明。因此,未来研究亟需解析去新生脂肪生成在代谢功能障碍相关脂肪性肝病自然病程各阶段的具体作用机制,并探索其作为联动肝脏与全身代谢健康的干预靶点之潜力,这将为最终构建代谢功能障碍相关脂肪性肝病的精准防治体系提供关键理论依据。 2.4.3 代谢功能障碍相关脂肪性肝病中脂肪组织功能障碍介导的机制 在代谢功能障碍相关脂肪性肝病的研究中,脂肪组织的功能障碍被认定为是一个重要的致病机制,其核心病理在于:功能失调的脂肪组织因胰岛素抵抗而脂解失控,导致过量游离脂肪酸涌入肝脏,成为肝内脂质异常蓄积的主要来源[59]。这一过程常由高脂、高糖饮食等因素触发,它们能引发脂肪组织应激、损害胰岛素信号传导,并减少有益脂肪因子的生成。 值得关注的是,脂肪组织功能障碍同样是体质量正常代谢功能障碍相关脂肪性肝病患者的重要致病机制之一[60],这类患者虽体质量指数正常,但常存在胰岛素抵抗及类似脂肪营养不良的特征,如臀部和大腿等部位脂肪含量显著减少,这揭示了脂肪分布异常与代谢风险之间的复杂关联,表明体质量正常并不能排除显著的代谢异常[61]。因此,后续研究应重点探讨脂肪组织功能障碍对代谢功能障碍相关脂肪性肝病发病的具体影响机制以及胰岛素抵抗和脂肪分布异常之间的关系。此外,严重的肝脂肪变性通常发生在遗传性、家族性和获得性脂肪营养不良患者中[62]。其中,获得性脂肪营养不良通常与自身免疫反应有关,并与代谢功能障碍相关脂肪性肝病的发病密切相关。值得注意的是,随着艾滋病治疗中抗逆转录病毒疗法及癌症免疫疗法的广泛应用,由此引发的获得性脂肪营养不良及其导致的严重代谢功能障碍相关脂肪性肝病并发症,正成为一个日益重要的临床课题[63]。临床实践中,瘦型代谢功能障碍相关脂肪性肝病常被忽视或误诊。根据2022年美国胃肠病学会的报告,7%-20%的代谢功能障碍相关脂肪性肝病患者体质量正常[8],这类患者通常年龄较大、以男性居多,其心血管代谢风险因素与超重或肥胖患者相当甚至更高,但对其独特的临床表现与疾病进展规律仍缺乏深入研究[8]。大规模流行病学分析为此提供了风险警示,一项涵盖94 181名代谢功能障碍相关脂肪性肝病患者的研究显示,瘦型患者的全因死亡风险明显高于非瘦型患者[64]。在肝脏特异性结局方面,尽管部分研究显示肝脏事件发生率与肥胖患者无显著差异[65],但一项长达20年的随访研究揭示,瘦型患者基线纤维化阶段虽较低,但其进展为严重肝病的长期风险却更高[66]。这些证据共同表明,瘦型代谢功能障碍相关脂肪性肝病患者同时面临着独特的肝脏进展风险和更高的全身性心血管代谢风险,并且这些风险独立于体质量变化。 综合而言,脂肪组织功能障碍是贯穿肥胖与瘦型代谢功能障碍相关脂肪性肝病的共同关键病理环节。针对瘦型代谢功能障碍相关脂肪性肝病这一特殊且高风险群体,未来的临床与研究重点应集中于建立更精准的早期识别体系、阐明其特有的发病机制,并制定有针对性的干预策略。 2.4.4 代谢功能障碍相关脂肪性肝病中临床表型介导的机制 在代谢功能障碍相关脂肪性肝病研究领域,异质性发病机制逐渐被关注和探讨,基于临床表型的风险分层有非常重要的应用价值[67]。由于代谢功能障碍相关脂肪性肝病的进展影响因素复杂,采用数据降维技术进行患者聚类分析已成为揭示其内在异质性的关键策略,该方法在肥胖及2型糖尿病研究中已得到广泛应用[68]。有研究对代谢功能障碍相关脂肪性肝病患者的血清代谢组数据进行深度分析,识别并划分出3种代谢亚型:A型(47%)、B型(27%)和C型(26%)[9]。 尽管这3种亚型在代谢功能障碍相关脂肪性肝病及纤维化患者的占比上相近,并且胰岛素抵抗与糖化血红蛋白A1c水平无显著差异,但A型患者的血清极低密度脂蛋白-甘油三酯水平显著低于B型和C型患者。值得注意的是,与B型、C型相比,A型患者的血清甘油三酯、胆固醇、极低密度脂蛋白、小而低密度脂蛋白胆固醇及残余脂蛋白胆固醇水平均较低,其心血管疾病10年风险评估值亦较低,并且Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸等代谢功能障碍相关脂肪性肝病风险等位基因频率更低[9]。在另一项美国第三次国家健康与营养调查数据的分析中,研究者基于腹部超声诊断脂肪肝,并结合2019年12月前的死亡数据开展两阶段聚类分析[69],该研究基于21个基线临床及实验室变量(如体质量指数、腰围、腰臀比、血红蛋白、糖化血红蛋白、尿酸、高密度脂蛋白胆固醇及胰岛素抵抗的稳态模型评估等),成功识别出3个不同群体,其中,较群体2(年长、肥胖、胰岛素抵抗显著)及群体3(年长、糖尿病患病率低但伴高血压和动脉粥样硬化性血脂异常)而言,群体1(较年轻、体质量正常、心血管代谢风险较低)的全因死亡率及心血管死亡率风险较高,但群体2与群体3在全因死亡率上无显著差异。值得注意的是,群体1中体质量正常、胰岛素敏感且血脂正常的代谢功能障碍相关脂肪性肝病患者是否主要与肝脏遗传成分相关,或在纤维化、肝硬化发生风险上是否存在差异,仍有待深入探究;同时,群体2的高胰岛素抵抗是否主要源于脂肪组织功能障碍,群体3的高血压及动脉粥样硬化性血脂异常是否由去新生脂肪生成增强所致,这些问题均尚未明确[70]。 另一项研究通过对甘油三酯及脂蛋白水平、糖化血红蛋白、总胆固醇/高密度脂蛋白胆固醇等参数分析,识别出5个代谢相关脂肪性肝病聚类,已在英国生物库数据库中成功验证[70]。聚类研究显示,不同群体在2型糖尿病、冠心病及全因死亡率风险上具有很大区别,其中被界定为严重胰岛素抵抗相关代谢相关脂肪性肝病的群体3显示生存结局较差,并且心血管代谢风险显著高于其他群体;其他聚类群体则分别与轻度肥胖伴血脂异常、年龄因素、脂蛋白水平等存在关联[70]。上述证明,代谢功能障碍相关脂肪性肝病患者的临床表型特征与不同聚类之间存在较强的依赖性,且其分布情况与所采用的临床参数密切相关。 综上所述,尽管现有聚类模型已初步揭示了代谢功能障碍相关脂肪性肝病患的异质性谱系,但该领域研究仍处于发展阶段,未来研究需针对聚类分析并精准结合代谢功能障碍相关脂肪性肝病患者肝脏表型,以保证准确性,并致力于在临床实践中构建更精准的患者分层标准体系。 2.4.5 代谢功能障碍相关脂肪性肝病中遗传变异代谢功能障碍介导的机制 基于遗传与代谢特征的交互模式,代谢功能障碍相关脂肪性肝病患者可被划分为“代谢驱动”与“遗传驱动”两种亚型[53]。代谢驱动群体的特点是代谢底物供应过多,还伴有外周脂解和去新生脂肪生成增强的情况;而遗传驱动群体则没有这些典型的代谢紊乱表现,反而存在明显的氧化还原失衡,具体表现为β-羟基丁酸与乙酰乙酸的比率升高,提示存在线粒体功能障碍[53]。这些发现提示遗传和代谢在代谢功能障碍相关脂肪性肝病发生中是相互作用的,但这两种因素具体是如何共同影响疾病不同表型的目前还未清楚。所以,后续研究应该深入探讨这两种因素在不同患者群体中的具体作用机制。 其他学者还发现还原应激会促进细胞应激和脂肪生成,这可能是代谢功能障碍相关脂肪性肝病的一个重要病理机制[71],表明代谢功能障碍相关脂肪性肝病的病理变化不仅与代谢功能障碍直接相关,也和细胞内氧化还原状态密切关联。CHEN等[72]根据多个基因变异分析将患者划分为7种代谢类别,并指出遗传变异的叠加效应在不同代谢表型中具有关键作用,证实了代谢功能障碍相关脂肪性肝病在个体之间存在遗传异质性。 临床中,患者特征分型是预测代谢功能障碍相关脂肪性肝病进展的重要手段。有研究人员通过6个常规临床特征(年龄、体质量指数、糖化血红蛋白等)归纳划分出两种代谢功能障碍相关脂肪性肝病类型。两种类型的临床发展轨迹不一样,其中,肝脏特异性聚类群体含有较多Patatin样磷脂酶域蛋白3的异亮氨酸148甲硫氨酸代谢功能障碍相关脂肪性肝病风险等位基因,肝脏病变进程较快,但心血管疾病风险比较低[73];而心血管代谢聚类群体的慢性肝病发生率和肝脏特异性群体相近,但血糖和甘油三酯水平更高,尽管其慢性肝病发生率与前者相近,但具有更高的心血管疾病和2型糖尿病的风险[73]。结果说明代谢功能障碍相关脂肪性肝病存在异质性,不同的代谢特征可能导致疾病不同的临床表征。此外,肝脏脂蛋白滞留也与代谢功能障碍相关脂肪性肝病的不同表型形成有密切关系。有研究通过构建的多基因风险评分模型发现两种不同的代谢功能障碍相关脂肪性肝病表型:一种是肝脏特异性表型,特征是肝脏脂质滞留,但循环中甘油三酯相对较低;另一种是系统性表型,伴有较高的心血管和代谢疾病风险,并且与严重肝病有关联[11]。总之,在代谢功能障碍相关脂肪性肝病的发病机制研究中,遗传与变异的影响以及代谢功能障碍本身的作用不容忽视,虽然相关研究进行了初步证据的提供,如不同表型在临床上的显著差异,但此方面诸多问题仍未解决。遗传变异对代谢路径的具体影响以及其导致出现代谢功能障碍相关脂肪性肝病不同表型,需通过更大规模和更多临床实践数据作支撑,并深入探究,所以,未来治疗方案的制定应先着重揭示机制间的内在联系。 2.5 代谢功能障碍相关脂肪性肝病共性机制与异质性因素的动态交互作用 2.5.1 分子基础——遗传异质性与共性代谢通路的交叉调控 遗传变异作为核心异质性因素,通过与代谢功能障碍相关脂肪性肝病共性代谢通路(如胰岛素信号、脂质转运)的精准交叉构建了疾病发生的分子基础,其中,PNPLA3 I148M和TM6SF2 E167K是当前研究最明确的遗传标志物,二者通过差异化调控共性通路,揭示了异质性因素对疾病进程的双向干预模式。 (1)PNPLA3 I148M:通过胰岛素抵抗通路放大脂质蓄积的正反馈循环。 Patatin样磷脂酶域包含蛋白3是调控肝脏脂质代谢的关键分子,其I148M变异(rs738409)是代谢功能障碍相关脂肪性肝病最强的遗传风险因素之一[74]。该变异并非直接触发肝脂肪变,而是通过增强共性机制中的胰岛素抵抗通路,形成“胰岛素抵抗-PNPLA3过表达-脂质蓄积”的病理性正反馈循环[75]。 在分子机制上,胰岛素通过激活磷脂酰肌醇3-激酶-蛋白激酶B信号通路信号通路上调固醇调节元件结合蛋白1c(SREBP-1c)的表达与核转位;而固醇调节元件结合蛋白1c可通过结合PNPLA3基因启动子区的固醇调节元件,直接激活其转录[76-77]。PNPLA3 I148M变异体与固醇调节元件结合蛋白1c的结合亲和力较野生型显著提高(约2.3倍),导致PNPLA3蛋白在肝细胞内的蓄积量增加;而变异型PNPLA3的磷脂酶活性显著降低,无法有效降解肝细胞内的甘油三酯前体,同时通过抑制脂肪甘油三酯脂酶的活性进一步减少甘油三酯分解,最终导致肝内脂质大量堆积[78-79]。临床研究证实,携带PNPLA3 I148M变异的代谢功能障碍相关脂肪性肝病患者胰岛素抵抗指数与肝脂肪含量呈显著正相关,并且该相关性独立于体质量指数[80],直接验证了遗传变异与共性代谢通路的协同致病效应。 (2)TM6SF2 E167K:通过抑制极低密度脂蛋白前体分泌通路加剧脂质输出障碍。 E167K变异导致TM6SF2蛋白的内质网滞留率增加(约40%),破坏了内质网-高尔基体间的脂质转运效率,使极低密度脂蛋白前体无法正常组装为成熟极低密度脂蛋白前体并分泌至血液[81]。临床研究显示,携带该变异的代谢功能障碍相关脂肪性肝病患者,其肝脏极低密度脂蛋白前体1(大颗粒极低密度脂蛋白前体)的分泌速率降低约35%,同时伴随肝内甘油三酯含量升高两三倍,患者通常保持相对正常的胰岛素敏感性[82]。更重要的是,这种分泌障碍与胰岛素抵抗通路存在协同作用:胰岛素抵抗本身可通过固醇调节元件结合蛋白1c上调肝脏甘油三酯合成,而TM6SF2变异进一步阻断甘油三酯的输出通路,形成“合成亢进-输出受阻”的双重打击,显著加速肝脂肪变向非酒精性脂肪肝炎的进展,并且严重胰岛素抵抗可进一步提高TM6SF2变异携带者的肝脂肪变风险[83]。这种“通路特异性”交互进一步体现了遗传异质性的调控多样性,也为后续精准干预提供了靶点依据。 2.5.2 表型异质性——瘦型与肥胖型代谢功能障碍相关脂肪性肝病的交互模式差异 (1)瘦型代谢功能障碍相关脂肪性肝病:脂肪组织功能障碍放大氧化应激与炎症损伤。 瘦型代谢功能障碍相关脂肪性肝病(体质量指数< 25 kg/m2)占全球代谢功能障碍相关脂肪性肝病患者的10%-30%,其核心异质性特征是脂肪组织功能障碍,而非脂肪量不足。尽管瘦型患者的白色脂肪组织总量正常,但脂肪细胞的分化成熟度降低,并且棕色脂肪组织的产热功能受损,导致脂肪组织对游离脂肪酸的储存能力下降,进而引发“异位脂肪沉积”(如肝脏、胰腺)[84]。 在分子层面,瘦型代谢功能障碍相关脂肪性肝病患者的脂肪组织存在显著的氧化应激失衡:脂肪细胞内环氧二十碳三烯酸的降解速率增加,导致环氧二十碳三烯酸/二羟基二十碳三烯酸比值降低(约0.6倍于健康人群),而该比值的下降与白色脂肪组织炎症及肝脂肪变呈显著负相关[84]。同时,瘦型患者的血清脂联素水平显著降低(通常< 7 μg/mL),较肥胖型患者低约40%[85]。脂联素作为脂肪组织分泌的关键抗炎激素,可通过激活腺苷酸活化蛋白激酶通路抑制肝细胞核因子κB炎症信号;而脂联素不足导致腺苷酸活化蛋白激酶通路激活不足,使相同程度的肝脂肪变引发更严重的肝细胞凋亡(caspase-3活性升高约2倍)及肝星状细胞活化[85]。因此,瘦型代谢功能障碍相关脂肪性肝病的交互核心是“脂肪组织功能障碍→氧化应激/炎症放大”,而非肥胖型的“脂肪量过剩→胰岛素抵抗”,这也解释了为何部分瘦型患者虽无明显代谢异常,却仍快速进展为肝纤维化。 (2) 肥胖型代谢功能障碍相关脂肪性肝病:脂肪细胞肥大强化胰岛素抵抗的“溢出效应”。 肥胖型代谢功能障碍相关脂肪性肝病(体质量指数≥30 kg/m2)的核心异质性因素是脂肪细胞肥大,其与共性机制的交互以“游离脂肪酸溢出-胰岛素抵抗”为核心。当白色脂肪组织容量超过储存极限时,脂肪细胞发生肥大(直径> 100 μm),导致脂肪组织缺氧及巨噬细胞浸润(CD11c?巨噬细胞比例升高约3倍),进而引发脂肪组织炎症[84]。 这种炎症状态通过两种方式强化胰岛素抵抗:①巨噬细胞分泌的肿瘤坏死因子、白细胞介素6等细胞因子抑制脂肪细胞胰岛素受体底物1的磷酸化,降低脂肪组织对胰岛素的敏感性;②肥大脂肪细胞的游离脂肪酸释放速率增加(约2.5倍),导致肝脏、肌肉等外周组织的游离脂肪酸摄入过多[84]。肝脏内过量游离脂肪酸通过激活 Toll 样受体4-核因子κB 通路引发炎症,同时通过甘油二酯-蛋白激酶Cε信号通路通路抑制肝细胞胰岛素信号,形成“脂肪细胞肥大→游离脂肪酸溢出→胰岛素抵抗→肝脂肪变”的恶性循环。与瘦型代谢功能障碍相关脂肪性肝病相比,肥胖型代谢功能障碍相关脂肪性肝病患者的疾病进展更依赖于“量效关系”(脂肪量→游离脂肪酸浓度→炎症程度),而异质性因素(如脂肪细胞大小、巨噬细胞浸润程度)则决定了这一关系的“效率”,例如,相同体质量指数的肥胖患者中,脂肪细胞肥大更显著者的肝纤维化风险升高约2.1倍,进一步印证了表型异质性的临床意义。 2.5.3 环境扰动——饮食、酒精与昼夜节律对交互网络的动态调控 外部环境因素(如高脂饮食、酒精、昼夜节律紊乱)作为“动态变量”,通过扰动“遗传-代谢”交互网络进一步重塑代谢功能障碍相关脂肪性肝病的疾病轨迹。分子病理流行病学研究证实,环境暴露与遗传/代谢异质性的交互是导致代谢功能障碍相关脂肪性肝病个体差异的重要原因,即相同环境因素对不同遗传背景或代谢状态的个体,可产生截然不同的疾病影响[86]。 肝脏脂质代谢具有严格的昼夜节律,受时钟基因/脑和肌肉芳香烃受体核转位蛋白样1异二聚体等核心节律基因调控,例如,白天(人类活动期)肝脏以甘油三酯合成为主,夜间则以β-氧化为主,这种节律平衡依赖于节律基因对代谢酶的时序性调控。高脂饮食可通过两种方式扰动这一节律:①直接下调肝脏Bmal1的表达(约50%),导致其靶基因的节律性表达消失,破坏甘油三酯合成与β-氧化的平衡[87];②激活下丘脑-垂体-肾上腺轴,使皮质醇水平升高,进一步抑制节律基因的转录。这种节律紊乱与遗传异质性存在显著协同作用,例如,携带PNPLA3 I148M变异的小鼠在高脂饮食喂养下,其肝脏PNPLA3的表达节律完全消失(野生型小鼠仍保持昼夜波动),并且肝甘油三酯蓄积量较野生型小鼠高约40%[87],这提示环境因素可通过“节律重塑”增强遗传异质性对共性通路的调控效应,也解释了为何长期昼夜颠倒(如轮班工作)的代谢功能障碍相关脂肪性肝病患者的疾病进展速度显著快于作息规律者。 酒精是代谢功能障碍相关脂肪性肝病进展的重要环境诱因,尤其当与代谢性饮食(如高糖高脂的MASH饮食)联合作用时,可通过激活“中性粒细胞外陷阱-NLRP3炎症小体-肝星状细胞”的级联反应打破“遗传-代谢”交互的稳态,加速肝纤维化。BABUTA等[88]的研究显示,短期代谢功能障碍相关脂肪性肝炎饮食(4周)联合每日酒精暴饮(5 g/kg)可使小鼠肝脏中性粒细胞外陷阱形成增加约3倍,同时激活NLRP3炎症小体,导致白细胞介素1β分泌升高4倍;而白细胞介素1β进一步通过旁分泌作用激活肝星状细胞,使α-平滑肌肌动蛋白(肝纤维化标志物)的表达升高5倍,并且这一效应在PNPLA3 I148M变异小鼠中更显著(α-平滑肌肌动蛋白表达额外升高30%)。从机制上看,酒精的有害作用源于其代谢产物乙醛对肝细胞的直接损伤,以及氧化应激的加剧,例如,CUBERO等[89]指出,酒精可通过激活Kupffer细胞的还原型烟酰胺腺嘌呤二核苷酸磷酸氧化酶产生大量活性氧,活性氧不仅直接损伤肝细胞线粒体,还可通过激活转化生长因子β/Smad通路进一步促进肝星状细胞向肌成纤维细胞转化。这种“酒精-代谢饮食-遗传变异”的三重交互构成了代谢功能障碍相关脂肪性肝病进展的“高危模型”,也为临床干预提供了“多靶点联合”的思路。"

| [1] LIM S, KIM JW, TARGHER G. Links between metabolic syndrome and metabolic dysfunction-associated fatty liver disease. Trends Endocrinol Metab. 2021;32(7):500-514. [2] KAHL S, STRAßBURGER K, PACINI G, et al. Dysglycemia and liver lipid content determine the relationship of insulin resistance with hepatic OXPHOS capacity in obesity. J Hepatol. 2025;82(3): 417-426. [3] SMITH GI, SHANKARAN M, YOSHINO M, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130(3):1453-1460. [4] SAITO T, TSUCHISHIMA M, TSUTSUMI M, et al. Molecular pathogenesis of metabolic dysfunction-associated steatotic liver disease, steatohepatitis, hepatic fibrosis and liver cirrhosis. J Cell Mol Med. 2024;28(12):e18491. [5] NÚÑEZ-SÁNCHEZ MÁ, MARTÍNEZ-SÁNCHEZ MA, MARTÍNEZ-MONTORO JI, et al. Lipidomic Analysis Reveals Alterations in Hepatic FA Profile Associated With MASLD Stage in Patients With Obesity. J Clin Endocrinol Metab. 2024;109(7):1781-1792. [6] LUUKKONEN PK, PORTHAN K, AHLHOLM N, et al. The PNPLA3 I148M variant increases ketogenesis and decreases hepatic de novo lipogenesis and mitochondrial function in humans. Cell Metab. 2023;35(11):1887-1896.e5. [7] WANG Y, HONG S, HUDSON H, et al. PNPLA3(148M) is a gain-of-function mutation that promotes hepatic steatosis by inhibiting ATGL-mediated triglyceride hydrolysis. J Hepatol. 2025;82(5):871-881. [8] LONG MT, NOUREDDIN M, LIM JK. AGA Clinical Practice Update: Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Lean Individuals: Expert Review. Gastroenterology. 2022;163(3):764-774.e1. [9] MARTÍNEZ-ARRANZ I, BRUZZONE C, NOUREDDIN M, et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology. 2022;76(4):1121-1134. [10] BERGHEIM I, MORENO-NAVARRETE JM. The relevance of intestinal barrier dysfunction, antimicrobial proteins and bacterial endotoxin in metabolic dysfunction-associated steatotic liver disease. Eur J Clin Invest. 2024;54(7):e14224. [11] JAMIALAHMADI O, DE VINCENTIS A, TAVAGLIONE F, et al. Partitioned polygenic risk scores identify distinct types of metabolic dysfunction-associated steatotic liver disease. Nat Med. 2024; 30(12):3614-3623. [12] GUREVITZ C, ROSENSON RS. Metabolic Dysfunction-Associated Steatotic Liver Disease, Hypertriglyceridemia and Cardiovascular Risk. Eur J Prev Cardiol. 2024. doi:10.1093/eurjpc/zwae388. [13] KONG J, XIE Y, FAN R, et al. Exercise orchestrates systemic metabolic and neuroimmune homeostasis via the brain-muscle-liver axis to slow down aging and neurodegeneration: a narrative review. Eur J Med Res. 2025;30(1):475. [14] DEVASIA AG, RAMASAMY A, LEO CH. Current Therapeutic Landscape for Metabolic Dysfunction-Associated Steatohepatitis. Int J Mol Sci. 2025; 26(4):1778. [15] YANAI H, ADACHI H, HAKOSHIMA M, et al. Metabolic-Dysfunction-Associated Steatotic Liver Disease—Its Pathophysiology, Association with Atherosclerosis and Cardiovascular Disease, and Treatments. Int J Mol Sci. 2023;24(20):15473. [16] ABDERRAHMANI A, YENGO L, CAIAZZO R, et al. Increased Hepatic PDGF-AA Signaling Mediates Liver Insulin Resistance in Obesity-Associated Type 2 Diabetes. Diabetes. 2018;67(7):1310-1321. [17] LIAO Y, CHEN Q, LIU L, et al. Amino acid is a major carbon source for hepatic lipogenesis. Cell Metab. 2024;36(11):2437-2448.e8. [18] YAO Z, GONG Y, CHEN W, et al. Upregulation of WDR6 drives hepatic de novo lipogenesis in insulin resistance in mice. Nat Metab. 2023;5(10):1706-1725. [19] COHEN CC, LI KW, ALAZRAKI AL, et al. Dietary sugar restriction reduces hepatic de novo lipogenesis in adolescent boys with fatty liver disease. J Clin Invest. 2021;131(24):e150996. [20] ALASMARI AA, AL-KHALIFAH AS, BAHAMMAM AS, et al. Ramadan fasting model exerts hepatoprotective, anti-obesity, and anti-hyperlipidemic effects in an experimentally-induced nonalcoholic fatty liver in rats. Saudi J Gastroenterol. 2024;30(1):53-62. [21] YOUNOSSI ZM, KOENIG AB, ABDELATIF D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73-84. [22] XIANG M, TIAN X, WANG H, et al. Inappropriate Diet Exacerbates Metabolic Dysfunction-Associated Steatotic Liver Disease via Abdominal Obesity. Nutrients. 2024;16(23):4208. [23] RAZA S, RAJAK S, UPADHYAY A, et al. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front Biosci (Landmark Ed). 2021;26(2):206-237. [24] ZHANG J, WANG Y, FAN M, et al. Reactive oxygen species regulation by NCF1 governs ferroptosis susceptibility of Kupffer cells to MASH. Cell Metab. 2024;36(8):1745-1763.e6. [25] XU X, KOBAYASHI S, QIAO W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116(7):1843-1852. [26] YOUNOSSI Z, ANSTEE QM, MARIETTI M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11-20. [27] WARIS G, AHSAN H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14. [28] BABU AF, CSADER S, MÄNNISTÖ V, et al. Effects of exercise on NAFLD using non-targeted metabolomics in adipose tissue, plasma, urine, and stool. Sci Rep. 2022;12(1):6485. [29] CHALASANI N, YOUNOSSI Z, LAVINE JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592-1609. [30] ZHU X, AIKEBAIER G, BAN X, et al. Increased serum β-hydroxybutyrate/acetoacetate ratio and aggravated histological liver inflammation in females with metabolic dysfunction-associated steatotic liver disease and polycystic ovary syndrome. J Mol Cell Biol. 2025;16(11):mjae048. [31] CHAN WK, CHUAH KH, RAJARAM RB, et al. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J Obes Metab Syndr. 2023;32(3):197-213. [32] SOTO A, SPONGBERG C, MARTININO A, et al. Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond. Biomedicines. 2024;12(2):397. [33] TAUIL RB, GOLONO PT, DE LIMA EP, et al. Metabolic-Associated Fatty Liver Disease: The Influence of Oxidative Stress, Inflammation, Mitochondrial Dysfunctions, and the Role of Polyphenols. Pharmaceuticals (Basel). 2024; 17(10):1354. [34] SVOBODOVÁ G, HORNÍ M, VELECKÁ E, et al. Metabolic dysfunction-associated steatotic liver disease-induced changes in the antioxidant system: a review. Arch Toxicol. 2025;99(1):1-22. [35] ALLAMEH A, NIAYESH-MEHR R, ALIARAB A, et al. Oxidative Stress in Liver Pathophysiology and Disease. Antioxidants (Basel). 2023;12(9):1653. [36] WOO BAIDAL JA, LAVINE JE. The intersection of nonalcoholic fatty liver disease and obesity. Sci Transl Med. 2016;8(323):323rv1. [37] MASCITELLI L, PEZZETTA F. Obesity, inflammation, and risk of atrial fibrillation or flutter. Am J Med. 2006;119(7):e9-e11. [38] CHEN Y, CHEN H, WANG Y, et al. LncRNA LINK-A Remodels Tissue Inflammatory Microenvironments to Promote Obesity. Adv Sci (Weinh). 2024;11(10):e2303341. [39] TACKE F, WEISKIRCHEN R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: mechanisms, treatment and prevention. Ann Transl Med. 2021;9(8):729. [40] QIN W, WENG J. Hepatocyte NLRP3 interacts with PKCε to drive hepatic insulin resistance and steatosis. Sci Bull (Beijing). 2023;68(13):1413-1429. [41] SUN L, WANG Y, BIAN F, et al. Bioinspired optical and electrical dual-responsive heart-on-a-chip for hormone testing. Sci Bull (Beijing). 2023; 68(9):938-945. [42] BITTENCOURT A, BRUM PO, RIBEIRO CT, et al. High fat diet-induced obesity causes a reduction in brain tyrosine hydroxylase levels and non-motor features in rats through metabolic dysfunction, neuroinflammation and oxidative stress. Nutr Neurosci. 2022;25(5):1026-1040. [43] SÁEZ-LARA MJ, ROBLES-SANCHEZ C, RUIZ-OJEDA FJ, et al. Effects of Probiotics and Synbiotics on Obesity, Insulin Resistance Syndrome, Type 2 Diabetes and Non-Alcoholic Fatty Liver Disease: A Review of Human Clinical Trials. Int J Mol Sci. 2016;17(6):928. [44] MOREIRA RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131(11): 1728-1734. [45] FABREGAT I, MORENO-CÀCERES J, SÁNCHEZ A, et al. TGF-β signalling and liver disease. FEBS J. 2016;283(12):2219-2232. [46] LIU YJ, KIMURA M, LI X, et al. ACMSD inhibition corrects fibrosis, inflammation, and DNA damage in MASLD/MASH. J Hepatol. 2025;82(2):174-188. [47] KOZLITINA J, SOOKOIAN S. Global Epidemiological Impact of PNPLA3 I148M on Liver Disease. Liver Int. 2025;45(3):e16123. [48] LAURIDSEN BK, STENDER S, KRISTENSEN TS, et al. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur Heart J. 2018;39(5):385-393. [49] LIU DJ, PELOSO GM, YU H, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49(12):1758-1766. [50] ROMEO S, SANYAL A, VALENTI L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020;31(1):35-45. [51] JOHNSON SM, BAO H, MCMAHON CE, et al. PNPLA3 is a triglyceride lipase that mobilizes polyunsaturated fatty acids to facilitate hepatic secretion of large-sized very low-density lipoprotein. Nat Commun. 2024;15(1):4847. [52] LUUKKONEN PK, ZHOU Y, SÄDEVIRTA S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167-1175. [53] LUUKKONEN PK, QADRI S, AHLHOLM N, et al. Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2022;76(3):526-535. [54] MUKHERJEE P, FUKUDA S, LUKMANTO D, et al. Label-free metabolic imaging of non-alcoholic-fatty-liver-disease (NAFLD) liver by volumetric dynamic optical coherence tomography. Biomed Opt Express. 2022;13(7):4071-4086. [55] DONNELLY KL, SMITH CI, SCHWARZENBERG SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005; 115(5):1343-1351. [56] LAMBERT JE, RAMOS-ROMAN MA, BROWNING JD, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726-735. [57] LAWITZ EJ, LI KW, NYANGAU E, et al. Elevated de novo lipogenesis, slow liver triglyceride turnover, and clinical correlations in nonalcoholic steatohepatitis patients. J Lipid Res. 2022;63(9): 100250. [58] AHMED A, CULE M, BELL JD, et al. Differing genetic variants associated with liver fat and their contrasting relationships with cardiovascular diseases and cancer. J Hepatol. 2024;81(6):921-929. [59] GASTALDELLI A, CUSI K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019;1(4):312-328. [60] STEFAN N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol. 2020;8(7):616-627. [61] STEFAN N, SCHICK F, HÄRING HU. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017;26(2):292-300. [62] LIM K, HAIDER A, ADAMS C, et al. Lipodistrophy: a paradigm for understanding the consequences of “overloading” adipose tissue. Physiol Rev. 2021; 101(3):907-993. [63] EIGENTLER T, LOMBERG D, MACHANN J, et al. Lipodystrophic Nonalcoholic Fatty Liver Disease Induced by Immune Checkpoint Blockade. Ann Intern Med. 2020;172(12):836-837. [64] WONGTRAKUL W, CHARATCHAROENWITTHAYA N, CHARATCHAROENWITTHAYA P. Lean non-alcoholic fatty liver disease and the risk of all-cause mortality: An updated meta-analysis. Ann Hepatol. 2024;29(3):101288. [65] YOUNES R, GOVAERE O, PETTA S, et al. Caucasian lean subjects with non-alcoholic fatty liver disease share long-term prognosis of non-lean: time for reappraisal of BMI-driven approach? J Hepatol. 2022;71(2):382-390. [66] HAGSTRÖM H, NASR P, EKSTEDT M, et al. Risk for development of severe liver disease in lean patients with nonalcoholic fatty liver disease: A long-term follow-up study. Hepatol Commun. 2017;2(1):48-57. [67] 邓彦.限时进食对代谢相关脂肪性肝病的影响及作用机制研究[D].武汉:武汉科技大学,2024. [68] STEFAN N, SCHULZE MB. Metabolic health and cardiometabolic risk clusters: implications for prediction, prevention, and treatment. Lancet Diabetes Endocrinol. 2023;11(6):426-440. [69] YI J, WANG L, GUO J, et al. Novel metabolic phenotypes for extrahepatic complication of nonalcoholic fatty liver disease. Hepatol Commun. 2023;7(1):e0016. [70] YE J, ZHUANG X, LI X, et al. Novel metabolic classification for extrahepatic complication of metabolic associated fatty liver disease: A data-driven cluster analysis with international validation. Metabolism. 2022;136:155294. [71] SINGH C, JIN B, SHRESTHA N, et al. ChREBP is activated by reductive stress and mediates GCKR-associated metabolic traits. Cell Metab. 2024;36(1):144-158.e7. [72] CHEN Y, DU X, KUPPA A, et al. Genome-wide association meta-analysis identifies 17 loci associated with nonalcoholic fatty liver disease. Nat Genet. 2023;55(10):1640-1650. [73] RAVERDY V, TAVAGLIONE F, CHATELAIN E, et al. Data-driven cluster analysis identifies distinct types of metabolic dysfunction-associated steatotic liver disease. Nat Med. 2024;30(12):3624-3633. [74] JEGODZINSKI L, RUDOLPH L, CASTVEN D, et al. PNPLA3 I148M variant links to adverse metabolic traits in MASLD during fasting and feeding. JHEP Rep. 2025;7(8):101450. [75] BRIL F, KALAVALAPALLI S, LOMONACO R, et al. Insulin resistance is an integral feature of MASLD even in the presence of PNPLA3 variants. JHEP Rep. 2024;6(7):101092. [76] LIANG H, XU J, XU F, et al. The SRE Motif in the Human PNPLA3 Promoter (-97 to -88 bp) Mediates Transactivational Effects of SREBP-1c. J Cell Physiol. 2015p;230(9):2224-2232. [77] DIF N, EUTHINE V, GONNET E, et al. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J. 2006;400(1):179-188. [78] ROMEO S, KOZLITINA J, XING C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008; 40(12):1461-1465. [79] XU R, TAO A, ZHANG S, et al. Association between patatin-like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: a HuGE review and meta-analysis. Sci Rep. 2015;5(1):9284. [80] KARAMFILOVA V, GATEVA A, ASSYOV Y, et al. PNPLA3 I148M polymorphism in patients with nonalcoholic fatty liver disease, obesity and prediabetes. J Gastrointestin Liver Dis. 2019; 28(4):433-438. [81] LIU J, GINSBERG HN, REYES-SOFFER G. Basic and translational evidence supporting the role of TM6SF2 in VLDL metabolism. Curr Opin Lipidol. 2024;35(3):157-161. [82] ZHOU Y, LLAURADÓ G, OREŠIČ M, et al. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J Hepatol. 2015;62(3): 657-663. [83] BORÉN J, ADIELS M, BJÖRNSON E, et al. Effects of TM6SF2 E167K on hepatic lipid and very low-density lipoprotein metabolism in humans. JCI Insight. 2020;5(24):e144079. [84] HATELEY C, OLONA A, HALLIDAY L, et al. Multi-tissue profiling of oxylipins reveal a conserved up-regulation of epoxide: diol ratio that associates with white adipose tissue inflammation and liver steatosis in obesity. EBioMedicine. 2024;103: 105127. [85] DOBRE MZ, VIRGOLICI B, CIOARCĂ-NEDELCU R. Lipid Hormones at the Intersection of Metabolic Imbalances and Endocrine Disorders. Curr Issues Mol Biol. 2025;47(7):565. [86] HAMADA T, KEUM NN, NISHIHARA R, et al. Molecular pathological epidemiology: new developing frontiers of big data science to study etiologies and pathogenesis. J Gastroenterol. 2017; 52(3):265-275. [87] TUREK FW, JOSHU C, KOHSAKA A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308(5724):1043-1045. [88] BABUTA M, NAGESH PT, DATTA AA, et al. Combined insults of a MASH diet and alcohol binges activate intercellular communication and neutrophil recruitment via the NLRP3-IL-1β Axis in the liver. Cells. 2024;13(11):960. [89] CUBERO FJ, URTASUN R, NIETO N. Alcohol and liver fibrosis. Semin Liver Dis. 2009;29(2):211-221. |
| [1] | Gao Zengjie, , Pu Xiang, Li Lailai, Chai Yihui, Huang Hua, Qin Yu. Increased risk of osteoporotic pathological fractures associated with sterol esters: evidence from IEU-GWAS and FinnGen databases [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1302-1310. |
| [2] | Li Zijing, Chen Xuwu, Ouyang Xinye, Wang Maoyuan. Mitophagy impairment mediated muscular atrophy: insights from the Drosophila model [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(23): 5897-5905. |
| [3] | Han Jie, Hu Tianfa, Wu Yachao, Nong Bin, Yu Kailong. Forkhead box transcription factor O3 affects bone metabolism and participates in the pathological processes of various bone-related diseases [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(22): 5770-5781. |
| [4] | Yu Yueyue, Zhang Xu, Liu Yiwei, Meng Zihan, Hao Xinyue, Tian Chunyu, Li Ji’an, Zhang Yixin . Lipid types and knee osteoarthritis: a genome-wide association study in European populations [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(18): 4763-4770. |
| [5] | Zhou Zigui, Liu Jingjing. Association between triglyceride-glucose-body mass index and the risk of hypertension in middle-aged and elderly Chinese population [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(16): 4137-4145. |
| [6] | Huang Rui, Wang Jie, Zhang Ye, Liao Jian. Association between peri-implantitis and cytokine gene polymorphisms [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(14): 3745-3752. |
| [7] | Yao Lijuan, Wang Yinfeng, Ma Zhennan, Chen Leqin. Exercise-induced extracellular vesicles: action and mechanisms in occurrence and development of insulin resistance [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(13): 3412-3423. |
| [8] | Wang Peng, Lu Huan, Liu Haifeng, Li Feng. Mechanism by which andrographolide intervenes in insulin resistance in rats with gestational diabetes mellitus [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(12): 3058-3065. |
| [9] | Wang Shijie, Hu Xiaoyu, Duan Zhuoran, Tang Yingfeng, Wang Wei . Association between grip strength to weight ratio and new-onset cardiovascular and cerebrovascular diseases: a big data analysis of the China Health and Retirement Longitudinal Study [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(12): 3190-3197. |
| [10] | Liu Shuhong, Xie Yuhan, Huang Chengcheng, Yang Zhenguo, Li Yang. The relationship between gout and some lifestyles, cardiovascular disease, bone disease, and psychiatric disease [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(10): 2525-2535. |
| [11] | Cai Zhixing, Xia Qiufang, Chen Lili, Zhu Danyang, Zhu Huiwen, Sun Yanan, Liang Wenyu, Zhao Heqian. Effect of Roujishuncuiyin on the improvement of skeletal muscle insulin resistance in a mouse model of type 2 diabetes mellitus [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(35): 7537-7543. |
| [12] | Liu Xuan, Ding Yuqing, Xia Ruohan, Wang Xianwang, Hu Shujuan. Exercise prevention and treatment of insulin resistance: role and molecular mechanism of Keap1/nuclear factor erythroid2-related factor 2 signaling pathway [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(35): 7578-7588. |
| [13] | Guo Haizhen, Cong Zidong, Zhao Yuke, Li Xiaofeng, Yu Lu, Qian Shule, Wang Runying, Du Wuxun. Development of patch clamp technology in the past 10 years: visual analysis based on CiteSpace and VOSviewer [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(31): 6717-6726. |
| [14] | Peng Zehong, Zhu Xi, Wen Jianglong, Zhu Wenzhuo, Liu Chao, Tang Jianwei, Cao Ziyue, Zhu Lili. Causal relationship between 39 plasma coagulation factors and chronic kidney disease based on samples from the GWAS Catalog database [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(24): 5272-5280. |
| [15] | Chen Chunlan, Ye Meiyi, Pan Yuwei, Yuan Jia, Zhou Pengjun. Immunomodulatory effect of umbilical cord mesenchymal stem cells on type 2 diabetes mellitus [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(23): 5031-5040. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||