Chinese Journal of Tissue Engineering Research ›› 2021, Vol. 25 ›› Issue (2): 311-316.doi: 10.3969/j.issn.2095-4344.2951
Previous Articles Next Articles
Cell autophagy, pathway, regulation and its multiple correlations with pulmonary hypertension
Chen Xinling, Wang Shenglan
- Qinghai University, Xining 810016, Qinghai Province, China
-
Received:2020-01-07Revised:2020-01-15Accepted:2020-03-18Online:2021-01-18Published:2020-11-21 -
Contact:Wang Shenglan, Professor, Qinghai University, Xining 810016, Qinghai Province, China -
About author:Chen Xinling, Master candidate, Qinghai University, Xining 810016, Qinghai Province, China -
Supported by:the National Natural Science Foundation of China, No. 81860076
CLC Number:
Cite this article
Chen Xinling, Wang Shenglan. Cell autophagy, pathway, regulation and its multiple correlations with pulmonary hypertension[J]. Chinese Journal of Tissue Engineering Research, 2021, 25(2): 311-316.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
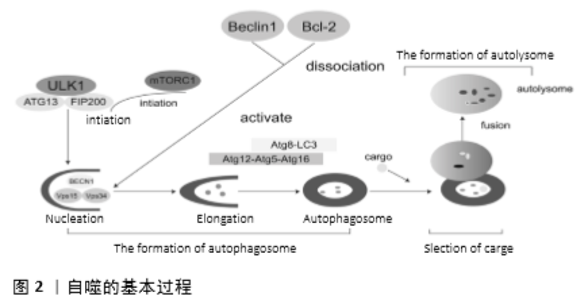
2.1 自噬的基本过程 2.1.1 自噬小体的形成 ①起始: 酵母中自噬小体形成的诱导受Atg1-Atg13-Atg17- Atg31-Atg29激酶(哺乳动物ULK1/2-ATG13-RB1CC1/FIP200)复合物的调控。在营养丰富的条件下,哺乳动物中的mTORC1会与ULK1/2-ATG13-RB1CC1/FIP200复合物结合并使ULK1/2和ATG13磷酸化,使ULK1/2和ATG13失活。然而,当细胞被雷帕霉素处理或缺乏营养时,mTORC1从复合物中解离,导致这些位点去磷酸化并诱导大自噬[7]。②成核:第Ⅲ类的PI3K(酵母Vps34)复合物的激活促进囊泡成核,该复合物由Atg6(哺乳动物Beclin-1)、Atg14(哺乳动物mATG14)、液泡分选蛋白34和15(Vps34和Vps15)的囊泡组成。其中Vps34、Vps15和BECN1蛋白构成囊泡的核心,而Vps34作用于PI(磷脂酰肌醇),产生PI3P,用于自噬体的成核和生长[8]。且第Ⅲ类的PI3K(酵母Vps34)复合物各组成成分均可以调节Vps34,从而调节自噬[9]。因此,在正常生理条件下,Bcl-2通过与Beclin1的BH3结构域结合抑制大自噬。在细胞饥饿期间,Beclin1通过Bcl-2或Beclin1的磷酸化,以及其他Bcl-2家族蛋白(tBid,Bad和BNIP3)或其他Beclin1结合蛋白(HMGB1,UVRAG,或Atg14L)竞争结合Beclin1的BH3结构域来实现Beclin1和Bcl-2的解离。Beclin1和Bcl-2解离以后,会促进Vps34的激活,后者激活第Ⅲ类的PI3K复合物,最终促进囊泡成核。③延伸和成熟:在酵母和哺乳动物中,囊泡的延伸需要Atg12-Atg5-Atg16和Atg8/LC3这2个泛素样结合系统的参与。Atg12-Atg5-Atg16L复合物可作为E3连接酶参与PE与LC3-Ⅰ的偶联以产生LC3-Ⅱ,LC3-Ⅱ可以与自噬体结合,在延伸的过程中发挥重要作用。同时,该复合物中的Atg16会发生二聚化并与自噬体结合,促进膜扩张。 2.1.2 货物的选择 自噬体膜内外表面的LC3B-Ⅱ与组成性表达的适配器分子p62/SQSTM1的相互作用与货物的选择密切相关。p62/SQSTM1包含一个泛素结合结构域用来隔离泛素化蛋白,而LC3B-Ⅱ相互作用区域(LIR结构域)则用于输送隔离蛋白。同时,另一种泛素结合蛋白ALFY与PI3P、p62和Atg5结合,以确保自噬体内存在蛋白质。而且,NBR1也参与自噬货物的选择,其包含2个LIR结构域和一个泛素结合结构域,会以不依赖p62的方式招募蛋白质聚集体。 2.1.3 自噬溶酶体的形成 PLEΚHM1作为Rab7效应蛋白,通过HOPS复合物和LC3/GABARAP蛋白调节自噬体和溶酶体的融合。HOPS复合物再通过与自噬体的SNARE相互作用来介导自噬体和溶酶体的融合。同时,STX17、VPS33A和Vps16结合,协助STX17发挥沉淀HOPS成分的作用。这个过程由镶嵌蛋白SNARES介导。近来发现,NFN2因子、早衰素、TECPR1、肌动蛋白VI和TOM1,以及DRAM1也有助于促进自噬体和溶酶体的融合[1],这两者的融合会促进货物的降解,见图2。"
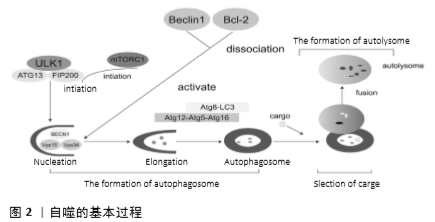

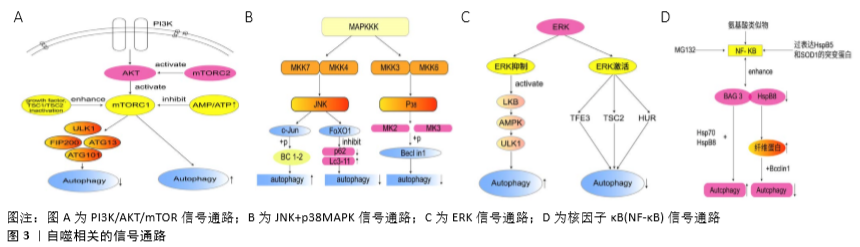
2.2 自噬相关的信号通路 2.2.1 PI3Κ/AΚT/mTOR信号通路 研究表明,PI3Κ/AΚT/mTOR信号通路参与调节细胞自噬,且不同的信号分子调节细胞自噬的机制也不同。作为PI3Κ/AΚT/mTOR信号通路中上游信号分子PI3K因其亚型不同,调节细胞自噬的机制也大相径庭,如Ⅰ类PI3K/Akt信号通过生长因子受体在癌细胞中激活,抑制自噬;相反,激活Ⅲ类PI3K却促进自噬。此外,磷酸酶和紧张素同源物(PTEN)去磷酸化并抑制Ⅰ类PI3K,从而抑制Akt活性并允许启动自噬。例如,熊果酸通过上调PTEN的表达从而抑制PI3K/Akt/mTOR通路来增强自噬。 AKT作为PI3K的下游底物,在各种肿瘤中经常高表达,它可以通过磷酸化Beclin 1刺激Beclin 1与14-3-3蛋白的相互作用,从而抑制自噬功能,也可以通过激活mTOR抑制自噬[10-11]。众所周知,mTOR是AKT的下游底物,是构成mTORC1和mTORC2的主要组分之一。据报道,细胞自噬主要是通过mTORC1来调控,mTORC2则是通过激活AKT间接促进mTORC1激活调控细胞自噬[12]。而mTORC1 主要受蛋白复合物TSC1/TSC2的活性调控。据称TSC1和TSC2蛋白会形成具有GTPase激活蛋白(GAP)活性的功能性肿瘤抑制复合物(GAP)。当TSC2与TSC1结合时,TSC2往往是作为一种小G蛋白(称为Rheb)的间隙发挥作用,将Rheb还原为不活跃的GDP结合形式并关闭mTORC1,当生长因子刺激和TSC1/TSC2失活时,Rheb被转化为活跃的GTP结合状态,从而增强mTORC1的激活,mTORC1的激活后可能通过抑制ULK1-FIP200-ATG13-ATG101来抑制自噬。而在营养不足的情况下,细胞ATP水平下降且AMP水平依次增加时,AMPΚ会被激活,此后通过一系列磷酸化事件刺激TSC1/TSC2的活性并抑制mTORC1,促进细胞自噬。mTORC2是通过Akt-Ser473磷酸化作用于Akt上游和Akt,从而能够调节mTORC1的活性,进而调节细胞自噬[10,12]。见图3A。 2.2.2 MAPK信号通路 MAPΚ信号通路调节细胞生长、细胞发育、分化和死亡,其信号转导是通过MAPΚΚΚ、MAPΚΚ和MAPΚ的顺序磷酸化发生的。目前在哺乳动物中鉴定6个MAPΚ亚家族:JNΚ1/2/3,ERΚ1/2,p38MAPΚ(p38α/β/γ/δ),ERΚ7/8,ERΚ3/4和ERΚ5/BMΚ1[3]。近期的研究表明JNΚ、p38MAPΚ、ERΚ信号通路参与调节细胞自噬。 (1)JNK信号通路:JNΚs可以被多种应激因素激活,包括紫外线辐射和氧化应激等。这些JNKs激活后可以诱导细胞凋亡和抑制细胞生长。JNΚ通路的上游激酶MΚΚ4和MΚΚ7(两者都是MAPKΚ)分别被不同的上游MAPKKΚ激活,一旦激活,JNΚs从细胞质转移到核。JNΚ含有大量的细胞核和细胞质靶基因,其中大多数是转录因子,包括AP-1(激活蛋白1)家族成员Jun、Fos和转录因子叉头框蛋白O(FoxO)[6]。JNK参与细胞自噬可能是通过以下2种机制:①JNK促进Bcl-2/Bcl-xL的磷酸化,促使Beclin1从Beclin1-Bcl-2/Bcl-xL复合体中分离[13];②JNK可能和ATG基因的表达有关。 研究发现第一种机制和自噬的调节密切相关。c-Jun在JNΚ介导的磷酸化后转位到细胞核。当c-Jun激活时,JNΚ在c-Jun的N末端磷酸化Ser63和Ser73以此激活c-Jun并增强其转录活性[14]。c-Jun激活后会促进Bcl-2/xL的表达及其磷酸化,进而影响和Beclin1结合的能力。当Bcl-2/xL磷酸化以后,促使Beclin-1从Beclin-1-Bcl-2/xL复合体中分离,促进Beclin1-hVps34-PI3Κ复合物的形成和Beclin1相关的PI3ΚⅢ的活性,进而诱导自噬[15]。 而且,大量的研究发现自噬的调节也与第二种机制密切相关。果蝇中进行的研究表明,经过氧化应激处理后,Atg1和Atg18的表达水平瞬间升高,并与自噬事件发生前JNΚ的峰值激活相一致,说明ATG基因是JNK通路的靶基因。众所周知,dFoxO蛋白是FoxO转录因子家族的成员,它可以激活JNΚ通路的下游。不少学者发现氧化应激因素通过小GTP酶Ral活性的JNΚ磷酸化FoxO4,并且诱导FoxO4核定位及增加其活性。然而,JNΚ作用的磷酸化位点是非保守性的FoxO1,FoxO3,FoxO6,表明FoxO转录因子直接控制启动和维持自噬。因此,有人提出细胞质FoxO转录因子是诱导自噬所必需的。据报道FoxO1的下调抑制p62降解 和LC3Ⅱ积累,导致自噬体减少。FoxO3在饥饿诱导的自噬中也是极其关键的,因为它调节ATG基因的转录,包括LC3和BNIP3。也有报道,FoxO1和Atg7相互作用对于自噬诱导是非常重要的。这些证据都说明FoxO在哺乳动物中诱导ATG基因表达[16]。见图3B。 (2)p38MAPK信号通路:据悉丝裂原激活的蛋白激酶激酶3和6(MΚΚ3和MΚΚ6)磷酸化后激活p38。而丝裂原激活的蛋白激酶激酶又根据刺激因素不同由不同的MAPΚΚΚ激活。MAPΚΚΚs的上游则是来自Rho亚家族(Rac1,Cdc242,Rho)和Ras样Rit亚家族的小GTPase(小GTP酶)。当膜受体刺激耦合到Rho/Ras信号传导时,会激活MΚΚs和p38[17]。在一项对Hela细胞自噬的研究中发现p38激活以后,会激活MAPΚAPΚ2(MΚ2)和MAPΚAPΚ3(MΚ3),然后通过磷酸化必需的ATG蛋白Beclin1,在Ser90处正向调节饥饿诱导的自噬。MAPΚAPΚ3,也被称为MΚ3,是唯一对Beclin1肽底物具有显著激酶活性的底物。MΚ2最初被发现为ERΚ1/2活化蛋白激酶、磷酸化HSP25和HSP27,后来发现可以被p38激活。据有关报道MΚ2和MΚ3在体外直接磷酸化Beclin1的Ser90,并且培养的细胞中MΚ2样激酶活性也介导饥饿诱导的Beclin1的Ser90磷酸化。总之,这些发现说明MΚ2/MΚ3可以介导Beclin1的Ser90磷酸化,而Beclin1的Ser90磷酸化会抑制自噬[18]。此外,学者发现除了MΚ2/MΚ3可以介导Beclin1的磷酸化,AKT [19]、Mst1[20]、ULK1[21]、ROCΚ1以及受体酪氨酸激酶表皮生长因子受体(EGFR)均可以介导Beclin1的磷酸化[22-23],从而在不同程度上调节自噬。见图3B。 其他的研究结果表明,在对促炎信号的响应中,p38MAPΚ直接磷酸化ULΚ1,抑制其激酶活性,破坏其与ATG13的功能复合物,并减少自噬。这些结果表明ULΚ1和ATG13的磷酸化已经成为调节ULΚ1激酶活性和自噬的关键机制[24]。也有报道指出星形胶质细胞的(GFAP)纤维酸性蛋白积累后能够激活p38,p38进而降低mTOR磷酸化,诱导细胞自噬[25]。但最近的一份报告表明,p38介导小胶质细胞的激活,通过依赖磷酸化抑制ULK1来减少自噬[24]。因此,在小胶质细胞和星形胶质细胞中,自噬的作用是相反的。此外,还观察到肝细胞内的低渗条件激活p38,P38的下游底物抑制自噬[26]。在HEK293细胞中p38α的缺失会导致自噬减少[27],而在小鼠胚胎成纤维细胞中它的激活则会抑制自噬小体-溶酶体的融合[28]。这些差异可能是由进行研究的细胞环境引起的。因此,p38介导的自噬调节的方向可能因细胞类型的不同而不同[25]。 (3)ERK信号通路:ERK信号通路参与细胞自噬的调节,可能是通过以下2种机制:①ERK抑制后导致ULK1-AMPK-LKB1轴的激活,调节细胞自噬;②ERK激活后作用于下游底物TFE3、TSC2或HuR,调节细胞自噬。 在第一种机制中,研究者发现发现抑制KRAS→RAF→ MEK→ERK信号会引起自噬,而ERK抑制MEK→ERK信号转导通路导致pS428LKB1磷酸化降低,AMPK (pT172)和ULK1(pS555)磷酸化增加,最终导致自噬的关键调节因子LKB1→AMPK→ULK1信号轴的激活,从而激活自噬。而ERK1/2可以通过丝氨酸428(pS428)的磷酸化抑制LKB1,LKB1反过来作用于 AMPK→ULK1/ATG1信号轴的上游,调节自噬[29]。 在第二种机制中学者发现细胞核中 MIT/TFE转录因子,特别是转录因子EB(TFEB)和转录因子E3 (TFE3) 调控大多数与溶酶体功能相关的Atg基因的表达。当细胞处于饥饿状态或溶酶体应激时,非磷酸化的TFEB/TFE3转位到细胞核,诱导自噬-溶酶体相关基因的表达。而细胞外信号调节激酶(ERK)被证明在S142使TFEB磷酸化,并调节其核转位,从而调节细胞自噬。有报道指出ERK在PLX4720诱导的BRAFV600黑色素瘤细胞的TFEB从胞浆到胞核转位中起主要作用,而且BRAFV600E下游效应蛋白ERK对TFEB的组成性磷酸化导致TFEB在胞质滞留和自噬溶酶体靶基因表达受损[30]。除此之外,有研究表明ERK磷酸化TSC2来阻断TSC1-TSC2复合物,从而刺激Rheb和mTORC1的活性。短暂或适度的ERK激活通过抑制mTORC1或mTORC2增加Beclin1来增加细胞保护性自噬,然而,持续或强烈的ERK激活完全抑制mTORC1和mTORC2,导致Beclin1的爆发性增加,导致最终的细胞毒性自噬。 最近的研究表明,靶向ERK通路可以调节自噬通量,例如ERK抑制剂或ERK沉默通过降低LC3-Ⅱ的表达和增加SQSTM1/p62的表达来干扰自噬。在营养丰富的条件下,通过mTOR介导的ULK1磷酸化抑制ULK1复合体[31]。也有报道指出在不同细胞中蛋白酶体抑制剂MG132决定ERK1/2的激活,ERK1/2调节HuR的细胞质转位,AMPK抑制有利于细胞质HuR(人类抗原R)的增加和p62蛋白的上调。而p62的mRNA作为Hur的靶标,可以影响细胞的自噬水平。但也有文献记载AMPK通过磷酸化HuR介导的转运蛋白,有利于HuR入核。在一项用人视网膜色素上皮细胞系ARPE-19的研究中,作者发现5-氨基咪唑-4-羧酰胺核糖核苷AICAR和蛋白酶体抑制剂MG132共处理可激活ERK1/2,介导HUR胞质增加、HUR磷酸化和p62蛋白上调。而单独使用AMPK抑制剂则有利于HuR的胞浆积聚,p62增加。因而作者假设AICAR 和MG132共处理触发ERK1/2激活,进而抑制AMPK,最终导致Hur细胞质积累和p62增加,从而影响细胞的自噬水平[32]。见图3C。 2.2.3 核因子κB细胞信号通路 核因子κB是细胞应激反应的关键转录因子,而核因子κB抑制剂IκBα可与核因子κB亚基结合形成IκBα/p50/p65复合物,该复合物隐藏核因子κB的核定位位点并阻止核因子κB易位进入细胞核,使得核因子κB处于失活状态,因此,核因子κB的激活需要其与IκB亚基的解离[33],研究者发现热应激以及不同的蛋白质聚集应激可以通过非典型途径激活核因子κB转录因子,诱导自噬过程。常见的处理主要是氨基酸类似物、MG132[34],以及过表达HspB5和SOD1的突变蛋白的处理。 在这些应激因素下,核因子κB激活并增强了BAG3和HspB8蛋白的表达,这些蛋白重新定位到绝大多数蛋白质聚集体,激活选择性自噬同时导致了蛋白质聚集体的清除。研究者还发现,蛋白质聚集应激因素激活核因子κB会增强BAG3和HspB8的表达,活化的核因子κB反过来又增强了BAG3和HspB8的表达。BAG3辅助伴侣蛋白是一种分子开关,将蛋白质定位于蛋白酶体或自噬降解蛋白质,同时增加自噬活性。BAG3与Hsp70和HspB8等伴侣蛋白结合,使其能够识别和结合错误折叠的蛋白质。据大量报道,BAG3有助于错误折叠的蛋白到mTOC的逆向转运,可以形成聚集体,并且通过它结合到Atg3促进自噬以及通过它和p62相互作用而促进选择性自噬[35]。因此,BAG3/HspB8复合物选择性地激活聚集蛋白如Htt43Q和SOD1G93A的自噬去除。研究者还观察到氨基酸类似物或MG132处理以及HspB5R120G、SOD1G85R和SOD1G93A的表达诱导BAG3和HspB8水平增加,并且刺激了自噬通量。HspB8还会伴随着波形纤维蛋白的增加,这些波形纤维蛋白通过与Beclin1相互作用抑制自噬[36]。 除了以上的热应激以及不同的蛋白质聚集应激可以激活核因子κB转录因子,进而诱导自噬过程。转化生长因子β1也可以激活核因子κB,转化生长因子β1诱导IκBα降解,导致核因子κB的增强。转化生长因子β1通过活化激酶TAΚ1和IκB激酶来促进核因子κB的激活。反过来,核因子κB和Smad3都可以增加Smad7的表达,Smad7竞争转化生长因子β1受体并下调转化生长因子β1/Smad3途径,提供负反馈有报道自噬与转化生长因子β1/Smad3途径密切相关[37],由此转化生长因子β1/Smad3途径可导致Beclin1的转录[38]。有报道表明抗凋亡蛋白XIAP也可以介导核因子κB激活。虽然详细机制尚不完全清楚,但现在已经发现XIAP可以通过RING和BIR1结构域之间的相互作用形成二聚体,这会导致转化生长因子β活化激酶1(TAΚ1)适配器蛋白的结合。TAΚ1被招募到该复合物中,并促进其二聚化和随后的激活,触发核因子κB信号通路。并且,XIAP的过表达会增强p65对Beclin1转录激活的扩增,最终诱导自噬[39],见图3D。 2.3 自噬与肺动脉高压 脉动脉高压是一种由阻力肺动脉的血管细胞过度增殖引起的疾病,其特征是肺血管重构和血管收缩,导致肺血管阻力增加,继而导致右心室肥厚和心力衰竭[40-41]。近年来的报道说明自噬和肺动脉高压的发生,发展密切相关。有研究发现ATG5靶向siRNA直接破坏自噬抑制大鼠肺动脉平滑肌细胞的增殖,并且必需的自噬基因Atg5-12的基因缺失可以抑制血清诱导的肺动脉平滑肌细胞的增殖并促进凋亡。但因其在肺动脉高压模型大鼠中激活自噬的方式不同,涉及的机制也有所不同。目前有AMPK介导的mTOR自噬信号通路、依赖于BNIP3的Beclin-1自噬信号通路、核因子κB信号通路均参与野百合碱MCT诱导的肺动脉高压大鼠的细胞自噬。 有报道指出抑制AMPK可显著减少心肌细胞自噬,而药物抑制AMPK可降低心肌细胞存活率,提示对依赖AMPK的心肌细胞自噬有保护作用,这些发现共同提示在代偿期自噬对心肌肥厚中的保护作用。而在野百合碱MCT处理2周和4周的大鼠中发现右心室磷酸化mTOR表达下调,磷酸化的AMPKα表达增加,但MCT处理6周的大鼠右心室磷酸化mTOR表达上调,磷酸化的AMPKα下降,除此之外,发现mTOR下游分子p70S6K,与观察到的磷酸化mTOR的变化相一致,当将AMPK抑制后自噬显著减少,这说明AMPK介导的mTOR自噬信号通路参与调节肺动脉高压大鼠的细胞自噬。另外发现右心室衰竭时BNIP3和Beclin1的过度表达则可以激活自噬,而HIF-1α作为BNIP3的上游分子靶点却不完全平行于BNIP3的改变,这表明除了依赖于BNIP3的Beclin1自噬信号通路可能还存在其他未被识别的上游分子通路,参与BNIP3调控的右心室重塑[41]。又有报道指出在野百合碱(MCT)诱导的肺动脉高压的大鼠模型中Belin1、LC3A、LC3B、LC3B/LC3A的比值增高,同时大鼠的肺裂解液中细胞核内核因子κB p65蛋白水平也增加,但胞质内核因子κB p65蛋白水平下降。然而核因子κB抑制剂PDTC可降低MTC大鼠细胞的自噬蛋白和核内核因子κB p65的水平,升高胞浆核因子κB p65蛋白水平,这些结果显示核因子κB激活后可以诱导MCT所诱导的肺动脉高压的大鼠的细胞自噬。目前已经发现在PAH动物模型中,mTOR抑制剂雷帕霉素治疗可以预防右室肥大和功能障碍,这说明雷帕霉素不仅可以改善肺血管重塑和右心室-肺动脉偶合,而且可能对自噬调节有直接作用,从而保护右心室[42]。 虽然目前已经报道几种肺动脉高压相关的自噬信号通路,但肺动脉高压的细胞自噬的分子机制目前还尚未阐述清楚。因此,研究肺动脉高压相关的细胞自噬的分子机制也是未来研究肺动脉高压的关键。"
| [1] RAVANAN P, SRIKUMAR IF, TALWAR P. Autophagy: the spotlight for cellular stress responses.Life Sci.2017:188(1): 53-67. [2] MIZUSHIMA N,KOMATSU M.Autophagy: Renovation of Cells and Tissues.Cell. 2011;147(4):728-741. [3] Jeanne MP, Vindis Cécile. Autophagy in health and disease: focus on the cardiovascular system.Essays Biochem.2017;61(6):721-732. [4] TASSET I, CUERVO AM. Role of chaperone-mediated autophagy in metabolism.FEBS J.2016;283(13):2403-2413. [5] STEVE C, PAULO P, HENRIQUE G. Molecular control of chaperone-mediated autophagy.Essays Biochem.2017;61(6):663-674. [6] ZHOU YY, LIY, JIANG WQ, et al. MAPK/JNK signalling: a potential autophagy regulation pathway.Biosci Rep.2015;35(3): e00199. [7] PARZYCH KR, KLIONSKY DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation.Antioxid Redox Signal.2014;20(3):460-473. [8] ESKELINEN EL. Autophagy: supporting cellular and organismal homeostasis by self-eating.Int J Biochem Cell Biol. 2019;111:1-10. [9] 吴葩,许彩民.Ⅲ型磷脂酰肌醇3-激酶复合物与自噬的研究进展[J].生物化学与生物物理进展,2012,39(3):210-216. [10] ZHAO GX, PAN H, OUYANG DY, et al. The critical molecular interconnections in regulating apoptosis and autophagy.Ann Med. 2015;47(4):305-315. [11] ZHANG J, ZHANG JX, ZHANG QL.PI3K/AKT/mTOR-mediated autophagy in the development of autism spectrum disorder.Brain Res Bull. 2016;125:152-158. [12] mo q, hu l, weng j, et al. Euptox A Induces G1 Arrest and Autophagy via p38 MAPK- and PI3K/Akt/mTOR-Mediated Pathways in Mouse Splenocytes.J Histochem Cytochem. 2017;65(9):543-558. [13] LUO S, RUBINSZTEIN DC.BCL2L11/BIM: a novel molecular link between autophagy and apoptosis.Autophagy.2013;9(1):104-105. [14] BOGOYEVITCH MA, KOBE B. Uses for JNK: the Many and Varied Substrates of the c-Jun N-Terminal Kinases.Microbiology Mol Bio Rev.2006;70(4):1061-1095. [15] WEI Y, SINHA SC, LEVINE B. Dual.Role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation.Autophagy.2008;4(7):949-951. [16] WU H, WANG MC, BOHMANN D. JNK protects Drosophila from oxidative stress by trancriptionally activating autophagy.Mech Dev.2011;126(8):624-637. [17] JULIA O, GIULIA F, PRAMSTALLER PP, et al. A new hypothesis for Parkinson’s disease pathogenesis:GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics.Mol Neurodegeneration.2018;13(1):40. [18] YONGJIE W, ZHENYI A, ZHONGJU Z, et al.The stress-responsive kinases MAPKAPK2/MAPKAPK3 activatetarvation-induced autophagy through Beclin 1 phosphorylation.Elife.2015;18(4):e05289. [19] WANG RC, WEI Y, AN Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation.Science.2012;338(6109): 956-959. [20] MAEJIMA Y, KYOI S, ZHAI P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2.Nat Med.2013;19(11):1478-1488. [21] RUSSELL RC, TIAN Y, YUAN H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase.Nat Cell Biol.2013;15(7):741-750. [22] GURKAR AU, CHU K, RAJ L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress.Nat Commun. 2013;4(1):2189. [23] CHENG G, LIU B, TANG Y, et al. Hypoxia-Induced Autophagy Promotes EGFR Loss in Specific Cell Contexts, Which Leads to Cell Death and Enhanced Radiosensitivity.Int J Biochem Cell Biol. 2019;111:12-18. [24] HE Y, SHE H, ZHANG T, et al. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1.J Cell Biol.2017; 217(1):315-328. [25] OBERGASTEIGER J, FRAPPORTI G, PRAMSTALLER PP, et al. A new hypothesis for Parkinson’s disease pathogenesis: GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics.Mol Neurodegener.2018;13(1):40. [26] PRICK T, THUMM M, HÄUSSINGER D, et al. Deletion of HOG1 leads to Osmosensitivity in starvation-induced, but not rapamycin-dependent Atg8 degradation and proteolysis: further evidence for different regulatory mechanisms in yeast autophagy.Autophagy.2006;2(3): 241-243. [27] WEBBER JL, TOOZE SA. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP.EMBO J. 2010;29(1):27-40. [28] KEIL E, HÖCKER R, SCHUSTER M, et al. Phosphorylation of Atg5 by the Gadd45 β–MEKK4-p38 pathway inhibits autophagy.Cell Death Differ. 2013;20(2):321-332. [29] KINSEY CG, CAMOLOTTO SA, BOESPFLUG AM, et al. Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers.Nat Med. 2019;25(4):620-627. [30] LI S, SONG Y, QUACH C, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance.Nature communications.2019;10(1):1-18. [31] KANG MJ, KIM SY, KWON EB, et al. Derrone induces autophagic cell death through induction of ROS and ERK in A549 cells.PloS one.2019;14(6):e0218659. [32] MARCHESI N, THONGON N, PASCALE A, et al. Autophagy Stimulus Promotes Early HuR Protein Activation and p62/SQSTM1 Protein Synthesis in ARPE-19 Cells by Triggering Erk1/2, p38MAPK, and JNK Kinase Pathways.Oxid Med Cell Longev. 2018;2018:4956080. [33] JIAO F, KAN C, YUJING X, et al. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-B and TGF-β1/Smad3 pathways.Drug Des Devel Ther.2018;12(1):1837-1853. [34] ZANOTTO-FILHO A, BRAGANHOL E, BATTASTINI AM, et al. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling.Invest New Drugs. 2012;30(6):2252-2262. [35] NIVON M, FORT L, MULLER P, et al. NFκB is a central regulator of protein quality control in response toprotein aggregation stresses via autophagy modulation.Mol Biol Cell.2016;27(11):1712-1727. [36] MINOIA M, BONCORAGLIO A, VINET J, et al. BAG3 induces the sequestration of proteasomal clients into cytoplasmic puncta: implications for a proteasome-to-autophagy switch.Autophagy.2014;10(9): 1603-1621. [37] FENG J, CHEN K, XIA Y, et al. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways.Drug Des Devel Ther. 2018;12:1837-1853. [38] LI J, CHEN K, LI S, et al. Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the TGF-β1/Smad pathway-mediated inhibition of extracellular matrix and autophagy.Drug Des Devel Ther. 2016;10:619-630 [39] LIN F, GHISLAT G, LUO S, et al. XIAP and cIAP1 amplifications induce Beclin 1-dependent autophagy through NFκB activation.Hum Mol Genet. 2015;24(10): 2899-2913. [40] CHEN R, JIANG M, LI BO, et al. The role of autophagy in pulmonary hypertension: a double-edge sword.Apoptosis.2018;23(9-10):459-469. [41] DENG Y, WU W, GUO S, et al. Altered mTOR and Beclin-1 mediated autophagic activation during right ventricular remodeling in monocrotaline-induced pulmonary hypertension.Respir Res. 2017;18(1):53. [42] ZHAI C, SHI W, FENG W, et al. Activation of AMPK prevents monocrotaline-induced pulmonary arterial hypertension by suppression of NF-κB-mediated autophagy activation.Life sciences.2018;208:87-95. [43] Fîlfan M, Sandu RE, Zăvăleanu AD, et al. Autophagy in aging and disease.Rom J Mor Emb.2017;58(1):27-31. [44] CHANG W, BAI J, TIAN S, et al. Autophagy protects gastric mucosal epithelial cells from ethanol-induced oxidative damage via mTOR signaling pathway.Exp Biol Med (Maywood). 2017;242(10):1025-1033. |
| [1] | Cui Xing, Sun Xiaoqi, Zheng Wei, Ma Dexin. Huangqin Decoction regulates autophagy to intervene with intestinal acute graft-versus-host disease in mice [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(7): 1057-1062. |
| [2] | Sun Youqiang, Ma Chao, Liang Mengmeng, Xin Pengfei, Zhang Hua, Xiang Xiaobing. The pivotal role of autophagy in bone cells: bone-related cell activity and bone metabolism [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(2): 276-282. |
| [3] | Yang Bingxuan, Jiang Tao, Jia Min, Li Peng, Liao Rongzhen, Li Zhao, Shao Min. Icariin promotes differentiation of mouse preosteoblasts via activating autophagy [J]. Chinese Journal of Tissue Engineering Research, 2022, 26(1): 64-69. |
| [4] | Ma Zetao, Zeng Hui, Wang Deli, Weng Jian, Feng Song. MicroRNA-138-5p regulates chondrocyte proliferation and autophagy [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(5): 674-678. |
| [5] | Xie Yang, Zhang Shujiang, Liu Menglan, Luo Ying, Yang Yang, Li Zuoxiao. Mechanism by which rapamycin protects spinal cord neurons in experimental autoimmune encephalomyelitis mice [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(5): 695-700. |
| [6] | Sun Mingshuai, Fan Chongshan, Li Kaijie, Gao Ruiyong, Wang Yudong, Shen Ruixue, Wang Lihe. Role of chondrocyte autophagy in osteoarthritis and its targeted therapy [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(35): 5688-5693. |
| [7] | Zhang Qunhui, Li Yimei, Zhang Dejun. Hypoxia-inducible factor and coronary heart disease: antagonism and protection [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(35): 5729-5734. |
| [8] | Zheng Weipeng, Hu Weijian, Zhao Guoyuan, Wei Hewei, Liu Zhijun, Wan Lei, Chen Sheng, Liao Zhihao. Effects of Bushen Jianpi Huoxue Recipe on cell proliferation and alkaline phosphatase activity in Beclin-1 overexpressing and silencing MC-3T3-E1 cells [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(29): 4650-4655. |
| [9] | Zhao Qianzeng, Zhao Zhenqun, Liu Wanlin. Endoplasmic reticulum stress regulates autophagy and apoptosis in steroid-induced avascular necrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(29): 4685-4690. |
| [10] | Yang Yuchen, Yang Peipei, Huang Biying, Zhang Qiang. Autophagy regulates osteoclast proliferation, differentiation and function through mitogen-activated protein kinase signaling pathway [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(26): 4192-4197. |
| [11] | Tong Jie, Liao Ying, Chen Zhengyu, Sun Guanghua . Osteoarthritic chondrocyte autophagy and regulation of mitogen-activated protein kinase signaling pathway [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(20): 3246-3251. |
| [12] | Li Songtao, Li Xinyi, Song Yunfeng, Ning Jiayin, Ren Qiang, Yang Renxu, Peng Bo. Maxing Xiongting Mixture regulates factors relevant to lung reshaping and vascular remodeling of hypoxic pulmonary hypertension rats [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(2): 274-280. |
| [13] | Jiang Tao, Ling Cuimin, Chen Qingzhen, Yang Bingxuan, Lin Yanping, Shao Min. Icariin prevents osteoporosis by activating autophagy and promoting osteoblast differentiation [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(17): 2643-2649. |
| [14] | Lai Shuaiwei, Zhang Shasha, Liu Xiaoyun, Haniya Mazhar, Amber Naz, He Lin, Zhu Hongxin. Ultraviolet resistance-associated gene (Uvrag) deficiency promotes cellular senescence in the heart [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(14): 2241-2246. |
| [15] | Zhang Xujian, Zhao Zhenqun, Liu Wanlin. The role of endoplasmic reticulum stress in the pathogenesis of steroid-induced avascular necrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2021, 25(11): 1759-1765. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||