Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (23): 5915-5924.doi: 10.12307/2026.334
Previous Articles Next Articles
Biological mechanisms and future research trends of cartilaginous endplate degeneration
Jiang Chao, Che Yanjun
- Orthopedics and Sports Medicine Center, Nanjing Medical University Affiliated Suzhou Hospital, Suzhou 215000, Jiangsu Province, China
-
Received:2025-04-03Accepted:2025-07-27Online:2026-08-18Published:2025-12-30 -
Contact:Che Yanjun, MD, Associate chief physician, Associate professor, Orthopedics and Sports Medicine Center, Nanjing Medical University Affiliated Suzhou Hospital, Suzhou 215000, Jiangsu Province, China -
About author:Jiang Chao, MS candidate, Orthopedics and Sports Medicine Center, Nanjing Medical University Affiliated Suzhou Hospital, Suzhou 215000, Jiangsu Province, China -
Supported by:Jiangsu Province Shuangchuang Talent Program Project, No. JSSCBS20211588 (to CYJ); Suzhou Gusu Health Talent Program Scientific Research Project, No. GSWS2021035 (to CYJ); Suzhou Multicenter Clinical Research Project for Major Diseases, No. DZXYJ202307 (to CYJ [project participant]); Suzhou Science and Technology Development Program (Healthcare Science and Technology Innovation), No. SKY2022185 (to CYJ); Suzhou City Combined Traditional Chinese and Western Medicine Research Fund Project, No. SKYD2023254 (to CYJ); Clinical Research Projects of Gusu College of Nanjing Medical University, Nos. GSKY20240208 (to CYJ) and GSKY20230402 (to CYJ [project participant]); Educational Research Project of Nanjing Medical University, No. 2023ZC081 (to CYJ)
CLC Number:
Cite this article
Jiang Chao, Che Yanjun. Biological mechanisms and future research trends of cartilaginous endplate degeneration[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(23): 5915-5924.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks

2.1 软骨终板退变研究进展的时间脉络 见表1。"


2.2 细胞凋亡在软骨终板退变中的作用 软骨终板退变的一个显著特征是软骨细胞凋亡[3]。细胞凋亡是一种基因控制的程序性细胞死亡过程,对维持生物体内环境稳定和组织器官正常生理功能至关重要[4]。细胞凋亡的触发可以通过内源性和外源性两种信号通路[5-6]。内源性信号通路与线粒体功能紧密相关,涉及Bcl-2家族蛋白的调控,这些蛋白调节线粒体外膜通透性,引发凋亡相关因子的释放,激活Caspase级联反应。外源性信号通路涉及死亡受体,如Fas细胞表面死亡受体和肿瘤坏死因子受体超家族成员,它们通过与相应配体结合激活下游信号通路,形成死亡诱导信号复合体,激活Caspase-8,触发细胞凋亡[7]。软骨细胞凋亡后,细胞核固缩、细胞皱缩、细胞膜起泡以及DNA片段化,细胞形态发生一系列变化,最终形成凋亡小体,被邻近细胞或巨噬细胞吞噬。 研究表明,核因子κB、Wnt、Hedgehog、丝裂原活化蛋白激酶、RhoA/Rock-1、蛋白激酶B /哺乳动物雷帕霉素靶蛋白、转化生长因子β和miRNA等信号通路在软骨终板细胞凋亡中发挥作用[3]。特别是半乳糖凝聚素3,作为β-半乳糖苷结合动物凝集素家族的一员,通过保守的碳水化合物识别结构域参与多种生理和病理过程[8-9]。刘岩路等[10]的实验发现,半乳糖凝聚素3抑制剂GB1107能够显著提高大鼠软骨终板细胞的凋亡率,表明半乳糖凝聚素3可能在软骨终板退变中具有调控作用。LI等[11]招募因椎间盘退变行腰椎手术患者,发现随着退变的进展,椎间盘软骨终板的软骨细胞数量和密度减少、胶原排列破裂并钙化,半乳糖凝聚素3、聚集蛋白聚糖和趋化因子(C-C基序)配体3的表达逐渐降低;在使用半乳糖凝聚素3抑制剂GB1107处理后,大鼠软骨终板细胞增殖显著减少、凋亡显著增加,并且细胞外基质代谢相关基因[基质金属蛋白酶3、趋化因子(C-C基序)配体3和聚集蛋白聚糖]的mRNA和蛋白表达水平低于对照组,这提示半乳糖凝聚素3可能是通过影响细胞外基质降解来影响椎间盘退变。目前针对半乳糖凝聚素33的研究较少,能否以细胞外基质降解为着力点来治疗椎间盘退变还是一个很大的挑战。 细胞凋亡通过复杂的信号通路网络影响软骨终板的健康,深入理解这些信号通路的调控机制对于开发抗凋亡治疗策略至关重要。未来的研究可以探索如何通过调节这些信号通路来抑制细胞凋亡,从而延缓软骨终板退变。 2.3 细胞衰老对软骨终板退变的作用 细胞衰老是指细胞在生理或病理条件下进入一种不可逆的生长停滞状态,但细胞并未死亡,而是保持代谢活性。细胞衰老过程涉及多种因素,包括基因表达的改变、细胞代谢失调、氧化应激增加以及细胞外基质的退化。椎间盘中的细胞衰老与软骨终板退变密切相关,表现为细胞外基质成分的改变、细胞代谢能力的下降以及生物力学特性的减弱。 2.3.1 DNA损伤与修复 DNA损伤是细胞衰老的核心机制之一,也是软骨终板退变的关键启动因素。随着年龄增长,细胞内DNA损伤逐渐累积、DNA修复能力下降,导致细胞功能障碍和组织退变。在软骨终板中,DNA损伤会抑制细胞的增殖能力,同时诱导细胞分泌促炎因子(如白细胞介素6、肿瘤坏死因子α),进一步加剧炎症反应和组织退变[2]。DNA损伤反应信号通路的激活是细胞衰老的关键因素之一,DNA损伤反应信号通路通过激活p53、p21等基因来抑制细胞周期,影响细胞正常代谢,导致细胞凋亡。 2.3.2 氧化应激 衰老过程中,细胞内活性氧水平升高,抗氧化防御机制减弱。氧化应激会损伤细胞内的蛋白质、脂质和DNA,导致细胞功能障碍。在软骨终板中,氧化应激会诱导细胞衰老,同时促进基质金属蛋白酶解整合素样金属蛋白酶的表达,加速细胞外基质的降解。研究表明,氧化应激通过激活核因子E2相关因子2信号通路来调节细胞抗氧化能力,但在衰老细胞中核因子E2相关因子2信号通路的活性显著下降[12]。另外,线粒体分裂是细胞代谢过程之一,线粒体的形态和功能发生改变导致线粒体膜电位下降,电子传递链功能受损,这会增加活性氧的产生,进而导致氧化应激[13]。基于这一发现,LIN等[7]通过靶向软骨终板细胞的外泌体递送核因子E2相关因子2以调节衰老软骨终板细胞线粒体分裂,有效缓解了软骨终板退变。 2.3.3 细胞分泌表型 衰老细胞会分泌多种细胞因子、趋化因子和蛋白酶,这种分泌表型被称为衰老相关分泌表型。衰老相关分泌表型因子会诱导周围细胞进入衰老状态,同时促进炎症反应和组织退变。一项对衰老髓核细胞的研究发现,随着椎间盘退变,衰老的髓核细胞数量显著增加,并且髓核细胞自噬受损[14],这有助于衰老相关分泌表型的上调;失调的衰老相关分泌表型破坏了髓核细胞的生存能力并启动了细胞外基质降解,相反,自噬的恢复通过抑制GATA结合蛋白4逆转衰老表型。 2.3.4 细胞代谢失调 衰老细胞的代谢功能发生改变,表现为糖酵解增加、线粒体功能下降,这种代谢失调会影响细胞的能量供应,削弱细胞的修复能力,与DNA损伤反应形成恶性循环。在软骨终板中,代谢失调会导致细胞外基质成分的改变,如Ⅱ型胶原蛋白和蛋白聚糖的减少;此外,细胞代谢失调还会导致细胞内酸性环境的改变,进一步影响细胞的正常功能。 细胞衰老是软骨终板退变的重要机制之一,涉及DNA损伤、氧化应激和细胞分泌表型的复杂相互作用。DNA损伤不仅抑制了细胞的增殖能力,还通过诱导促炎因子的分泌加剧了炎症反应。氧化应激则通过激活核因子E2相关因子2信号通路调节细胞的抗氧化能力,但在衰老细胞中这一信号通路的活性显著下降。目前对于软骨终板细胞衰老方面的研究较少,研究软骨终板细胞自噬的机制以及如何延缓细胞衰老,可能是治疗椎间盘退变的一种有前景的新策略。 2.4 炎症反应对软骨终板的影响 椎间盘退变的本质是慢性炎症,在退变椎间盘中可检测到高水平的促炎递质,主要是肿瘤坏死因子α和白细胞介素1β,并且促炎递质升高程度与椎间盘退变程度相关[15]。炎症反应导致椎间盘内代谢失衡,促进椎间盘退变。考虑到局部环境对细胞行为的显著影响,分析软骨终板在炎症条件下的潜在作用变得至关重要。研究发现,肿瘤坏死因子α和白细胞介素1β是参与椎间盘退变的两种主要炎症递质,体外暴露于人椎间盘细胞可引起诱导型一氧化氮合酶、一氧化氮、白细胞介素1β、白细胞介素6、白细胞介素8、白细胞介素20、前列腺素2和和肿瘤坏死因子α等炎症分子的上调,并伴随着分解代谢酶解整合素金属蛋白酶与硫蛋白和基质金属蛋白酶的上调,进而促进椎间盘退变的发生发展[16-17]。大规模高通量微阵列和蛋白质阵列分析显示,在白细胞介素1β处理后,软骨终板细胞群显示编码参与明胶、胶原蛋白、蛋白聚糖、层粘连蛋白、纤连蛋白和弹性蛋白降解的几种基质金属蛋白酶的基因表达上调,这几种降解酶会降解细胞外基质,而细胞外基质降解是椎间盘退变的主要标志,证明了白细胞介素1β营造的炎症环境增强了软骨终板细胞的分解代谢[16]。在模拟软骨终板微损伤的研究中发现,肿瘤坏死因子α对软骨终板细胞的影响较小,但会抑制细胞外基质合成,促进细胞外基质降解[18];向大鼠椎间盘内注射肿瘤坏死因子α相比注射PBS会引起更严重的椎体终板骨髓炎性改变,诱导软骨终板变性。综上所述,炎症因子会进一步刺激炎症反应的发生,促进软骨终板变性,但如何通过控制炎症反应来控制缓解这一过程还需深入研究。 炎症介导的细胞信号传导是一个复杂的过程,涉及多种细胞内信号通路。其中,经典Wnt信号通路(Wnt/β-catenin信号通路)、核因子κB及Yes1相关转录共激活因子/转录共激活因子同源物信号通路是细胞内3条重要信号通路,对于维持细胞的正常生理功能具有重要意义,这些信号通路之间存在复杂的交汇作用,广泛参与了细胞一系列生理及病理反应过程。 2.4.1 Wnt/β-catenin信号通路 Wnt蛋白与由Frizzled受体和低密度脂蛋白受体相关蛋白5/6组成的受体复合物结合时[19],Wnt信号通路被激活。在没有Wnt配体的情况下,细胞内磷酸化的β-catenin在骨架蛋白Axin和腺瘤性息肉病蛋白的作用下被激酶酪蛋白激酶Ⅰ和糖原合成酶激酶3β激活。磷酸化的β-catenin与β-转录激活因子重复序列蛋白C结合,导致其泛素化降解;当Wnt配体存在时,糖原合成酶激酶3β活性被抑制,β-catenin在细胞内积聚,在细胞核中β-catenin与T细胞因子/淋巴增强因子结合,激活靶基因启动子,从而启动Wnt信号通路下游靶基因的转录,表达细胞周期蛋白,如细胞周期蛋白D1、Runx2、骨特异性转录因子和基质金属蛋白酶[20-21],见图4。 鉴于Wnt信号通路的重要性,研究者们对此进行了深入研究[22-23]。例如,Dickkopf-1蛋白和骨硬化蛋白通过与成骨细胞表面的低密度脂蛋白受体相关蛋白5/6结合阻断Wnt信号"
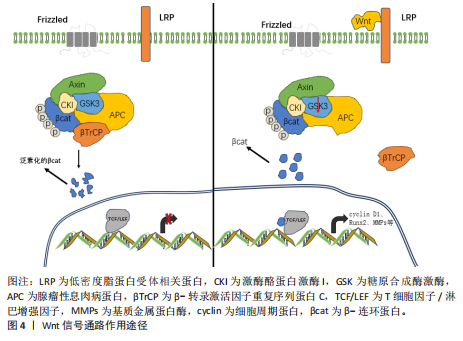

通路,抑制成骨细胞的增殖和分化、减少骨保护素的表达,从而增强核因子κB受体激活因子配体诱导破骨细胞分化的生物效应[24-25]。KROON等[23]将小鼠分为4组干预,A组皮下注射抗骨硬化蛋白抗体,B组皮下注射抗Dickkopf-1抗体,C组混合注射抗骨硬化蛋白/抗Dickkopf-1抗体(3∶1),D组为对照组,发现与对照组相比,A、B、C组椎间盘高度和β-catenin表达均升高,但只有A组椎间盘的机械性能增加;后续实验发现骨硬化蛋白的持续删除可通过上调Dickkopf-1得到补偿,证实了Wnt信号的补偿性抑制,认为抑制骨硬化蛋白或Dickkopf-1可以通过刺激Wnt信号来增强椎间盘结构,为开发治疗椎间盘退变的新疗法提供了潜在靶点。目前,抗骨硬化蛋白抗体已被临床用于绝经后妇女骨质疏松的治疗[22],但能否将抗骨硬化蛋白抗体大规模用于骨质疏松患者还是一个极具挑战性的难题。 2.4.2 核因子κB信号通路 在未激活状态下,核因子κB蛋白(通常是p50和p65亚基的二聚体)与抑制蛋白IκB结合在一起,被保留在细胞质中[26-27]。当细胞受到外界刺激,如细菌、病毒、应激、细胞因子(如肿瘤坏死因子α、白细胞介素1β)等,会激活核因子κB信号通路,激活的信号会通过一系列上游激酶最终激活IκB激酶复合体。IκB激酶复合体磷酸化IκB蛋白,导致其结构变化,磷酸化的IκB被泛素化[28],然后被蛋白酶体识别并降解。IκB降解后,核因子κB被释放,转移到细胞核中;在细胞核内,核因子κB与DNA上的特定序列(κB位点)结合,激活或抑制靶基因的转录。核因子κB激活基因表达后,新的IκB蛋白被合成,核因子κB再次与IκB结合被送回细胞质,等待下一次激活。核因子κB信号通路的异常激活与许多疾病有关,包括炎症性疾病、自身免疫疾病、癌症等,因此,核因子κB信号通路是许多药物开发的重要靶点。 骨桥蛋白由分泌性磷酸蛋白1基因编码,在骨骼和其他组织中高表达[29]。已经发现骨桥蛋白表达的异常水平与软骨终板退变密切相关,骨桥蛋白在退变软骨终板中的含量显著降低,并且呈时间依赖性[30]。骨桥蛋白是椎间盘微环境中的一个新线索,它与椎间盘的发展和退变有关。一项对退变和非退变椎间盘细胞的单细胞RNA测序分析提出,分泌性磷酸蛋白1是椎间盘的预测因子;当条件性敲除分泌性磷酸蛋白1后,骨桥蛋白缺失加重了与年龄相关的软骨终板退变,包括细胞外基质降解、细胞死亡、新生血管形成、破骨细胞活性和巨噬细胞侵袭,直接损害了椎间盘退变发展过程中软骨终板的稳态,表现在蛋白聚糖和Ⅱ型胶原蛋白的表达水平显著降低,表明细胞外基质合成代谢受损;基质金属蛋白酶13和X型胶原的表达增加,提示软骨终板发生了软骨细胞肥大和细胞外基质分解代谢[31]。于是,WANG等[31]进一步探索了骨桥蛋白缺失促进软骨终板退变的机制,发现骨桥蛋白缺失增强了软骨终板细胞中核因子κB信号、NLRP3和基质金属蛋白酶13的表达,但降低了蛋白聚糖的表达,而咖啡酸苯乙酯(核因子κB抑制剂)或核因子κB p65沉默处理可减轻或消除这些变化。 显然,骨桥蛋白缺失会增强核因子κB信号传导,进而促进软骨终板中的炎症反应。最近,一项联合分析揭示了不同细胞亚群中的基本致病因素会影响软骨终板稳态,其中骨桥蛋白是关键的[32]。因此,基于骨桥蛋白调控核因子κB也为椎间盘退变治疗提供了一条策略。 2.4.3 Yes1相关转录共激活因子/转录共激活因子同源物信号通路 Yes1相关转录共激活因子/转录共激活因子同源物信号通路,也称为Hippo信号通路,是一条在进化过程中高度保守的信号传导途径[33-35]。在正常生理状态下,Yes1相关转录共激活因子/转录共激活因子同源物信号通路通过一系列激酶级联反应最终调控下游效应分子Yes相关蛋白和转录共激活因子的活性。当信号通路激活时,Yes1相关转录共激活因子和转录共激活因子同源物会被磷酸化并滞留在细胞质中,从而无法进入细胞核内发挥作用;而当信号通路关闭时,未磷酸化的Yes1相关转录共激活因子/转录共激活因子同源物能够进入细胞核与TEAD家族转录因子结合,激活下游基因的表达,促进细胞增殖、抑制细胞凋亡[36]。 Yes1相关转录共激活因子/转录共激活因子信号通路在软骨细胞增殖和分化中发挥关键作用[37],会影响软骨终板的组成[31]。在健康软骨终板中,Yes1相关转录共激活因子/转录共激活因子途径有助于维持细胞更新和修复能力[38]。然而,在炎症条件下,Yes1相关转录共激活因子/转录共激活因子信号通路通过调节胶原和蛋白聚糖等基质分子的表达影响软骨终板的完整性,在软骨终板退变过程中,细胞外基质合成和降解的失衡是导致软骨终板功能障碍的关键因素[39]。另外,正如上文所述,炎症因子(如肿瘤坏死因子α和白细胞介素1β)通过核因子κB信号通路诱导基质降解酶,促进软骨降解;同时,炎症因子可通过转化生长因子β激活激酶1介导的磷酸化触发Yes1相关转录共激活因子/转录共激活因子降解。有意思的是,最近的研究发现,Yes1相关转录共激活因子/转录共激活因子通过阻止转化生长因子β激活激酶1底物的可及性和随后的IκB激酶α/β激活来拮抗核因子κB活性,Yes1相关转录共激活因子减弱核因子κB信号激活,并通过抑制基质降解酶的诱导来减少细胞外基质降解[40]。事实上,目前已经有许多核因子κB抑制剂(如糖皮质激素)在骨关节炎中发挥抗炎作用[41],由此可以设想,同时靶向Yes1相关转录共激活因子/转录共激活因子和核因子κB信号通路,或者更具体地说是Yes1相关转录共激活因子/转化生长因子β激活激酶1信号通路,可能会是椎间盘退变治疗更好结果的合理策略。 2.4.4 Piezo离子通道 Piezo1离子通道是一种阳离子通道蛋白,可以作为直接感受器。机械力通过引起细胞膜的形变激活Piezo通道,将机械刺激转换为电化学信号。脊椎动物有2种Piezo蛋白:Piezo1和Piezo2离子通道。机械刺激影响Piezo通道的阳离子通透性,对离子的通透性由大到小为:Ca2+ > K+> Na+> Mg2+,对Ca2+通透性的影响最大,对Mg2+通透性的影响最小。Piezo1比Piezo2对机械调节更敏感,Piezo2主要与触觉感知相关,包括轻柔触觉、本体感觉(如肌肉状态和平衡感知)以及内脏器官的机械变化(如血压、呼吸、膀胱充盈度)[35]。Piezo1广泛分布在人体不同组织中,其中在骨、软骨、骨骼肌、肌腱、椎间盘甚至其周围的结缔组织中稳定表达,参与大多数哺乳动物的各种生理过程,如淋巴管发育、轴突生长、血管发育、免疫调节和血压调节。因此,该文主要描述Piezo1离子通道。Piezo1存在于骨中的各种细胞中,包括骨髓间充质干细胞、成骨细胞、骨细胞、软骨细胞、内皮细胞等。Piezo1的表达受多种因素影响,在肢体发育过程中,Piezo1主要在分化的成骨细胞和软骨细胞中表达,而在破骨细胞中表达较少,并且Piezo1表达在出生后的小鼠中上调,但在皮质骨中随着年龄的增长而降低。除了衰老,机械刺激也可以影响Piezo1的表达,机械卸载降低Piezo1表达,而在适当机械刺激下Piezo1表达上调。 Piezo1在小鼠和人类软骨中均高表达。在软骨终板中,Piezo1直接感受机械应力,激活后诱导Ca2+内流进入细胞,从而启动下游信号通路的级联反应(如经典Wnt信号信号通路),参与细胞增殖、分化和基质的合成与降解;此外,Piezo1的激活伴随着下游炎症因子(肿瘤坏死因子α、白细胞介素1β)和分解代谢酶(基质金属蛋白酶、解整合素金属蛋白酶与硫蛋白)的表达上调。LI等[42]研究了Piezo1在骨细胞中促进骨合成代谢的机制,发现相较于对照组,缺失Piezo1基因小鼠表现为明显的骨缺损和低骨量,并且Wnt1信号降低。 Yoda1是Piezo1特异性变构激活剂,可降低通道激活所需阈值。GsMTX4是一种蜘蛛毒肽,抑制阳离子敏感机械通道,非特异性抑制Piezo1通道。在临床上,因过度机械应力导致患者椎间盘退变的情况很多,能否用Piezo1抑制剂减弱异常的机械传导,从而防止过度压力导致的细胞和基质受损,是一个潜在的治疗策略。为了解决骨质疏松给药不精确和不良反应大的问题,GUAN等[43]使用唑来膦酸盐装饰的聚乳酸-羟基乙酸复合物、超顺磁性氧化铁和Piezo1激动剂Yoda1制备了纳米载体,体内外实验显示该纳米载体可有效靶向骨质疏松性骨缺损部位并激活Piezo1通道,通过Yes1相关转录共激活因子/β-catenin信号轴改善成骨作用,促进成骨-血管生成耦合、显著加速骨缺陷内的骨重建,这一发现为治疗软骨终板退变提供了一种潜在的治疗方法。鉴于Piezo1通道在软骨终板退变的多方面作用,阐明Piezo1通道在软骨终板退变的精确机制,并思考如何调节并优化这些生理过程是未来研究的重点。 不同信号通路对软骨终板的影响总结,见表2。 炎症反应在软骨终板退变中发挥了关键作用,肿瘤坏死因子α和白细胞介素1β通过激活核因子κB和Wnt/β-catenin等信号通路诱导了基质金属蛋白酶的表达,加速了细胞外基质的降解。现有的抗炎治疗策略(如糖皮质激素)虽然能够缓解炎症反应,但长期使用可能导致不良反应。未来的研究可以探索如何通过靶向多个炎症信号通路(如核因子κB和Yes1相关转录共激活因子/转录共激活因子同源物)来更有效控制炎症反应,结合生物力学调控,开发更为精准的治疗方案。"


2.5 细胞外基质的变化及生物学意义 细胞外基质是软骨终板的重要组成部分[44],主要由多种大分子组成,包括胶原蛋白(尤其是Ⅱ型胶原蛋白)、蛋白聚糖(如聚集蛋白聚糖)以及非胶原蛋白(如弹性蛋白、纤维连接蛋白等)[3,45],这些成分共同为软骨终板提供了结构支持和生物学功能。Ⅱ型胶原蛋白是软骨终板中含量最为丰富的蛋白质,相较于Ⅰ型和Ⅲ型胶原,它对胶原酶具有显著的抵抗力。在生理状态下,Ⅱ型胶原蛋白保证了软骨终板承受高力和频繁渗透交换的能力,从而维持软骨终板的结构稳定和生理功能。 在软骨终板退变过程中,细胞外基质成分会发生显著变化:Ⅱ型胶原蛋白会被基质金属蛋白酶、解整合素样金属蛋白酶等酶分解[46],导致胶原蛋白网的破坏,伴随着Ⅰ型胶原的合成增多[47-49],同时蛋白聚糖的合成减少[50]、降解增加,导致基质的亲水性和弹性减弱,而椎间盘不同亚结构中的水含量取决于蛋白聚糖含量比值和施加的载荷,具有较高比例蛋白聚糖的区域表现出更高的水合水平,这是使软骨终板能够抵抗机械载荷的关键因素[51-52]。这些变化共同导致软骨终板的厚度减少、硬化和钙化。 细胞外基质成分的变化会直接影响软骨终板细胞的行为[53]。Ⅱ型胶原蛋白和蛋白聚糖的减少会减弱细胞与基质的相互作用,影响细胞的附着、增殖和分化[54-55];此外,还会改变细胞外的力学微环境,进而影响细胞的形态和功能[56]。不同的细胞外基质硬度对软骨终板细胞活性有不同影响[57-58]。基质硬化可能导致细胞凋亡和衰老,而基质钙化可能阻断营养物质的扩散,导致生物力学改变,进一步加剧退变过程[59-60]。 2.6 生物力学因素在软骨终板退变中的作用 软骨终板被认为是椎间盘的应力中心,承受着椎体的压应力、压缩力、拉伸力及液体流动的压力,柱状排列使其非常适合承受压应力,在脊柱中起到缓冲和允许一定程度活动的作用[51,61]。与其他软骨细胞一样,一定的压力对于软骨终板是至关重要的,有助于调节细胞代谢[62-63]。软骨终板细胞的代谢活动(包括蛋白多糖和胶原蛋白的合成与分解)受到力学负荷的影响,适当的力学刺激有助于维持椎间盘的健康,而不适当的负荷可能导致细胞活动失衡,与一些其他因素共同促进退变过程,见表3。 腰椎不稳会引起异常的力学刺激,导致软骨终板退变,进而发展为椎间盘退变,然而具体的机制还不明确。退变的软骨终板中会出现干酪样形态和感觉神经支配,引起中老年人腰背痛。LI团队[4]通过对异常应力下的软骨终板细胞进行转录组测序发现,Yes1相关转录共激活因子/转录共激活因子同源物信号通路是这一过程的关键,过表达Yes1相关转录共激活因子通过阻断趋化因子(C-C基序)配体3的启动子来抑制其转录,进而逆转终板重塑为干酪样形态;在大鼠手术部位局部注射腺相关病毒血清型5包裹的Yes1相关转录共激活因子过表达质粒,成功挽救了腰椎不稳手术诱导的软骨终板重塑,发现了软骨终板中Yes1相关转录共激活因子/转录共激活因子同源物信号通路诱导的破骨细胞基因集激活是治疗腰椎不稳引起的腰痛和椎间盘退变的潜在新靶点。 研究已证明,低张力牵引能更好地促进骨终板的主动重建,改善椎间盘的弹性模量和微纳米结构,因此,它进一步促进了椎间盘的再生和修复[39,64-66]。软骨终板结构的重塑保证了椎间盘的营养供应,可以延缓椎间盘的进一步退变。KANDEL等[67]证明营养因素和运输障碍是人类椎间盘恢复缓慢的关键因素。然而,椎间盘的生物力学微环境调节与细胞外基质协调的精确平衡尚未解决。 生物力学因素在软骨终板退变中发挥了重要作用,力学环境的改变不仅影响了细胞的代谢活动,还通过激活Piezo1和Yes1相关转录共激活因子/转录共激活因子同源物等信号通路加速了细胞外基质的降解。现有的力学调控策略(如固定牵引模型)虽然能够延缓退变,但临床应用仍面临挑战。探索如何通过智能材料或力学响应药物来优化力学微环境,并结合分子生物学手段,开发更为精准的治疗方案是未来主要的研究方向。 2.7 未来研究趋势 2.7.1 信号通路的深入探索与靶向治疗 未来的研究应进一步探索Wnt/β-catenin、核因子κB、Yes1相关转录共激活因子/转录共激活因子同源物等信号通路之间的相互作用及其在不同病理条件下的调控机制。例如,研究如何通过靶向这些信号通路来抑制炎症反应、细胞凋亡和基质降解,从而延缓软骨终板退变;此外,开发基于这些信号通路的小分子抑制剂或激活剂,在动物模型和临床试验中验证其疗效和安全性。 2.7.2 生物力学调控与材料相结合 未来研究需探索生物力学调控与再生材料的结合,以修复和重建软骨终板的结构与功能。例如:①开发力学响应再生材料,设计能够感知并响应力学刺激的智能材料,这些材料可以在特定的力学环境下释放生物活性因子或药物,促进软骨终板细胞的增殖和基质合成;②优化再生材料的力学设"


计,研究如何通过材料的力学特性(如弹性模量、孔隙率和渗透性)以及力学信号传导通路(如Piezo1、Yes1相关转录共激活因子/转录共激活因子同源物通路)来调控细胞行为。 2.7.3 人工智能与大数据的应用 在人工智能与AI技术快速发展的今天,研究者可以利用这些工具来分析软骨终板退变的复杂机制。例如,通过机器学习算法预测疾病进展,建立疾病预测模型,优化治疗方案,或从海量的临床数据中挖掘潜在的生物标志物和治疗靶点;此外,利用计算机模拟技术,通过有限元分析研究软骨终板在不同力学和生物化学条件下的行为,为实验设计和临床应用提供理论支持。"

| [1] FRANCISCO V, PINO J, GONZÁLEZ-GAY MÁ, et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat Rev Rheumatol. 2022;18(1):47-60. [2] SILWAL P, NGUYEN-THAI AM, MOHAMMAD HA, et al. Cellular Senescence in Intervertebral Disc Aging and Degeneration: Molecular Mechanisms and Potential Therapeutic Opportunities. Biomolecules. 2023;13(4):686. [3] ZEHRA U, TRYFONIDOU M, IATRIDIS JC, et al. Mechanisms and clinical implications of intervertebral disc calcification. Nat Rev Rheumatol. 2022;18(6):352-362. [4] LI H, TANG Y, LIU Z, et al. Lumbar instability remodels cartilage endplate to induce intervertebral disc degeneration by recruiting osteoclasts via Hippo-CCL3 signaling. Bone Res. 2024;12(1):34. [5] CAZZANELLI P, WUERTZ-KOZAK K. MicroRNAs in Intervertebral Disc Degeneration, Apoptosis, Inflammation, and Mechanobiology. Int J Mol Sci. 2020;21(10):3601. [6] WANG J, JING X, LIU X, et al. Naringin safeguards vertebral endplate chondrocytes from apoptosis and NLRP3 inflammasome activation through SIRT3-mediated mitophagy. Int Immunopharmacol. 2024;140:112801. [7] LIN Z, XU G, LU X, et al. Chondrocyte-targeted exosome-mediated delivery of Nrf2 alleviates cartilaginous endplate degeneration by modulating mitochondrial fission. J Nanobiotechnol. 2024;22(1):281. [8] CHEN X, YU C, LIU X, et al. Intracellular galectin-3 is a lipopolysaccharide sensor that promotes glycolysis through mTORC1 activation. Nat Commun. 2022;13(1):7578. [9] DONG R, ZHANG M, HU Q, et al. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int J Mol Med. 2018;41(2):599-614. [10] 刘岩路,胡 炜,艾克拜尔,等.抑制半乳糖凝集素3促进椎间盘软骨终板细胞凋亡诱导椎间盘退变[J]. 中国组织工程研究,2021,25(35):5599-5603. [11] LI J, HAN N, LIU Z, et al. Role of Galectin-3 in intervertebral disc degeneration: an experimental study. BMC Musculoskelet Disord. 2024;25(1):249. [12] ZHOU H, WU C, JIN Y, et al. Role of oxidative stress in mitochondrial dysfunction and their implications in intervertebral disc degeneration: Mechanisms and therapeutic strategies. J Orthop Translat. 2024;49:181-206. [13] WESTERMANN B. Mitochondrial fusion and fission in cell life and death. Nat RevMol Cell Biol. 2010;11(12):872-884. [14] WU O, JIN Y, ZHANG Z, et al. KMT2A regulates the autophagy-GATA4 axis through METTL3-mediated m6A modification of ATG4a to promote NPCs senescence and IVDD progression. Bone Res. 2024; 12(1):67. [15] KAPOOR M, MARTEL-PELLETIER J, LAJEUNESSE D, et al. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33-42. [16] DE LUCA P, DE GIROLAMO L, KOUROUPIS D, et al. Intervertebral disc and endplate cells response to IL-1β inflammatory cell priming and identification of molecular targets of tissue degeneration. Eur Cell Mater. 2020; 39:227-248. [17] XIA Q, ZHAO Y, DONG H, et al. Progress in the study of molecular mechanisms of intervertebral disc degeneration. Biomed Pharmacother. 2024;174:116593. [18] WANG D, LAI A, GANSAU J, et al. Lumbar endplate microfracture injury induces Modic-like changes, intervertebral disc degeneration and spinal cord sensitization - an in vivo rat model. Spine J. 2023;23(9):1375-1388. [19] JIA H, MA J, LV J, et al. Oestrogen and parathyroid hormone alleviate lumbar intervertebral disc degeneration in ovariectomized rats and enhance Wnt/β-catenin pathway activity. Sci Rep. 2016;6: 27521. [20] SMOLDERS LA, MEIJ BP, ONIS D, et al. Gene expression profiling of early intervertebral disc degeneration reveals a down-regulation of canonical Wnt signaling and caveolin-1 expression: implications for development of regenerative strategies. Arthritis Res Ther. 2013;15(1):R23. [21] 项 攀,车艳军,罗宗平.压应力激活SOST/Wnt/β-catenin通路诱导软骨终板细胞退变[J].中国组织工程研究,2025, 29(5):951-957. [22] GAO Y, CHEN N, FU Z, et al. Progress of Wnt Signaling Pathway in Osteoporosis. Biomolecules. 2023;13(3):483. [23] KROON T, BHADOURIA N, NIZIOLEK P, et al. Suppression of Sost/Sclerostin and Dickkopf-1 Augment Intervertebral Disc Structure in Mice. J Bone Miner Res. 2020; 37(6):1156-1169. [24] JIAO Z, CHAI H, WANG S, et al. SOST gene suppression stimulates osteocyte Wnt/β-catenin signaling to prevent bone resorption and attenuates particle-induced osteolysis. J Mol Med (Berl). 2023;101(5):607-620. [25] DIEGEL CR, KRAMER I, MOES C, et al. Inhibiting WNT secretion reduces high bone mass caused by Sost loss-of-function or gain-of-function mutations in Lrp5. Bone Res. 2023;11(1):47. [26] LI Q, VERMA IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002; 2(10): 725-734. [27] LAWRENCE T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. [28] YU H, LIN L, ZHANG Z, et al. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209. [29] SI J, WANG C, ZHANG D, et al. Osteopontin in Bone Metabolism and Bone Diseases. Med Sci Monit. 2020;26:e919159. [30] QU Y, WANG Y, WANG S, et al. A comprehensive analysis of single-cell RNA transcriptome reveals unique SPP1+ chondrocytes in human osteoarthritis. Comput Biol Med. 2023;160:106926. [31] WANG Y, ZHANG W, YANG Y, et al. Osteopontin deficiency promotes cartilaginous endplate degeneration by enhancing the NF-κB signaling to recruit macrophages and activate the NLRP3 inflammasome. Bone Res. 2024;12(1):53. [32] LI W, ZHANG S, ZHAO Y, et al. Revealing the Key MSCs Niches and Pathogenic Genes in Influencing CEP Homeostasis: A Conjoint Analysis of Single-Cell and WGCNA. Front Immunol. 2022;13:933721. [33] SUN K, GUO J, GUO Z, et al. The roles of the Hippo-YAP signalling pathway in Cartilage and Osteoarthritis. Ageing Res Rev. 2023;90:102015. [34] LI Z, LIN J, WU J, et al. The Hippo signalling pathway in bone homeostasis: Under the regulation of mechanics and aging. Cell Prolif. 2024;57(10):e13652. [35] ZHOU T, GAO B, FAN Y, et al. Piezo1/2 mediate mechanotransduction essential for bone formation through concerted activation of NFAT-YAP1-ß-catenin. Elife. 2020;9:e52779. [36] ZARKA M, HAŸ E, COHEN-SOLAL M. YAP/TAZ in Bone and Cartilage Biology. Front Cell Dev Biol. 2021;9:788773. [37] DING B, XIAO L, XU H. YAP1 controls degeneration of human cartilage chondrocytes in response to mechanical tension. Cell Biol Int. 2022;46(10):1637-1648. [38] ZHANG M, XIONG S, GAO D, et al. Tension regulates the cartilage phenotypic expression of endplate chondrocytes through the α-catenin/actin skeleton/Hippo pathway. J Cell Mol Med. 2024;28(4): e18133. [39] XIANG P, LUO ZP, CHE YJ. Insights into the mechanical microenvironment within the cartilaginous endplate: An emerging role in maintaining disc homeostasis and normal function. Heliyon. 2024;10(10):e31162. [40] DENG Y, LU J, LI W, et al. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat Commun. 2018;9(1):4564. [41] KINGSBURY SR, THARMANATHAN P, KEDING A, et al. Pain Reduction With Oral Methotrexate in Knee Osteoarthritis : A Randomized, Placebo-Controlled Clinical Trial. Ann Int Med. 2024;177(9):1145-1156. [42] LI X, HAN L, NOOKAEW I, et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. ELife. 2019;8:e49631. [43] GUAN H, WANG W, JIANG Z, et al. Magnetic Aggregation‐Induced Bone‐Targeting Nanocarrier with Effects of Piezo1 Activation and Osteogenic–Angiogenic Coupling for Osteoporotic Bone Repair. Adv Mater. 2024; 36(13):2312081. [44] SIVAN SS, HAYES AJ, WACHTEL E, et al. Biochemical composition and turnover of the extracellular matrix of the normal and degenerate intervertebral disc. Eur Spine J. 2014;23 Suppl 3:S344-353. [45] MA Z, LIU X, ZHANG M, et al. Research Progress on the Role of Cartilage Endplate in Intervertebral Disc Degeneration. Cell Biochem Funct. 2024;42(7):e4118. [46] KUHN A, RIEGGER J, TEIXEIRA GQ, et al. Terminal Complement Activation Is Induced by Factors Released from Endplate Tissue of Disc Degeneration Patients and Stimulates Expression of Catabolic Enzymes in Annulus Fibrosus Cells. Cells. 2023;12(6):887. [47] MOORE RJ. The vertebral endplate: disc degeneration, disc regeneration. Eur Spine J. 2006;15 Suppl 3(Suppl 3):S333-337. [48] LI W, NIU Y, QIU Z, et al. New evidence on the controversy over the correlation between vertebral osteoporosis and intervertebral disc degeneration: a systematic review of relevant animal studies. Eur Spine J. 2024;33(6):2354-2379. [49] DUDLI S, FIELDS AJ, SAMARTZIS D, et al. Pathobiology of Modic changes. Eur Spine J. 2016;25(11):3723-3734. [50] WEN ZQ, LIN J, XIE WQ, et al. Insights into the underlying pathogenesis and therapeutic potential of endoplasmic reticulum stress in degenerative musculoskeletal diseases. Mil Med Res. 2023;10(1):54. [51] ASHTON-MILLER JA, SCHULTZ AB. Biomechanics of the human spine and trunk. Exerc Sport Sci Rev. 1988;16:169-204. [52] CRUMP KB, ALMINNAWI A, BERMUDEZ-LEKERIKA P, et al. Cartilaginous endplates: A comprehensive review on a neglected structure in intervertebral disc research. JOR Spine. 2023;6(4): e1294. [53] BERMUDEZ-LEKERIKA P, CRUMP KB, TSERANIDOU S, et al. Immuno-Modulatory Effects of Intervertebral Disc Cells. Front Cell Dev Biol. 2022;10:924692. [54] GE Y, CHEN Y, GUO C, et al. Pyroptosis and Intervertebral Disc Degeneration: Mechanistic Insights and Therapeutic Implications. J Inflamm Res. 2022;15:5857-5871. [55] BIBBY SR, JONES DA, LEE RB, et al. The pathophysiology of the intervertebral disc. Joint Bone Spine. 2001;68(6):537-542. [56] NEWELL N, LITTLE JP, CHRISTOU A, et al. Biomechanics of the human intervertebral disc: A review of testing techniques and results. J Mech Behav Biomed Mater. 2017;69:420-434. [57] WANG N, TYTELL JD, INGBER DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10(1):75-82. [58] LI Z, YUE M, LIU X, et al. The PCK2-glycolysis axis assists three-dimensional-stiffness maintaining stem cell osteogenesis. Bioact Mater. 2022;18:492-506. [59] EMPERE M, WANG X, PREIN C, et al. Aggrecan governs intervertebral discs development by providing critical mechanical cues of the extracellular matrix. Front Bioeng Biotechnol. 2023;11:1128587. [60] HABIB M, HUSSIEN S, JEON O, et al. Intradiscal treatment of the cartilage endplate for improving solute transport and disc nutrition. Front Bioeng Biotechnol. 2023;11:1111356. [61] FERGUSON SJ, STEFFEN T. Biomechanics of the aging spine. Eur Spine J. 2003;12 Suppl 2(Suppl 2):S97-S103. [62] WALKER MH, ANDERSON DG. Molecular basis of intervertebral disc degeneration. Spine J. 2004;4(6 Suppl):158S-166S. [63] ADAMS MA. Biomechanics of back pain. Acupunct Med. 2004;22(4):178-188. [64] CHE YJ, GUO JB, HAO YF, et al. Regenerating and repairing degenerative intervertebral discs by regulating the micro/ nano environment of degenerative bony endplates based on low-tension mechanics. BMC Musculoskelet Disord. 2022;23(1):462. [65] CHE YJ, HOU JJ, GUO JB, et al. Low energy extracorporeal shock wave therapy combined with low tension traction can better reshape the microenvironment in degenerated intervertebral disc regeneration and repair. Spine J. 2021;21(1): 160-177. [66] CHE YJ, GUO JB, LIANG T, et al. Controlled immobilization-traction based on intervertebral stability is conducive to the regeneration or repair of the degenerative disc. Spine J. 2019;19(5):920-930. [67] KANDEL R, ROBERTS S, URBAN JPG. Tissue engineering and the intervertebral disc: the challenges. Eur Spine J. 2008;17 Suppl 4(Suppl 4):480-491. [68] LI C, BAI Q, LAI Y, et al. Advances and Prospects in Biomaterials for Intervertebral Disk Regeneration. Front Bioeng Biotechnol. 2021;9:766087. |
| [1] | Zhang Xianxu, Ma Zhong, Liu Xin, Huang Lei, Shen Wenxiang, Luo Zhiqiang . Lumbar fusion combined with unilateral fixation for lumbar degenerative diseases: biomechanics, technical evolution, and clinical applications [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(9): 2334-2342. |
| [2] | Zhu Xiaolong, Zhang Wei, Yang Yang. Visualization analysis of research hotspots and cutting-edge information in the field of intervertebral disc regeneration and repair [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(9): 2391-2402. |
| [3] | Liu Anting, Lu Jiangtao, Zhang Wenjie, He Ling, Tang Zongsheng, Chen Xiaoling. Regulation of AMP-activated protein kinase by platelet lysate inhibits cadmium-induced neuronal apoptosis [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1800-1807. |
| [4] | Cai Ziming, Yu Qinghe, Ma Pengfei, Zhang Xin, Zhou Longqian, Zhang Chongyang, Lin Wenping. Heme oxygenase-1 alleviates lipopolysaccharide-induced inflammatory response in nucleus pulposus mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1624-1631. |
| [5] | Yuan Xiaoshuang, Yang Xu, Yang Bo, Chen Xiaoxu, Tian Ting, Wang Feiqing, Li Yanju, Liu Yang, Yang Wenxiu. Effect of conditioned medium of diffuse large B-cell lymphoma cells on proliferation and apoptosis of human bone marrow mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1632-1640. |
| [6] | He Jiale, Huang Xi, Dong Hongfei, Chen Lang, Zhong Fangyu, Li Xianhui. Acellular dermal matrix combined with adipose-derived stem cell exosomes promotes burn wound healing [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1699-1710. |
| [7] | Chen Yulin, He Yingying, Hu Kai, Chen Zhifan, Nie Sha Meng Yanhui, Li Runzhen, Zhang Xiaoduo , Li Yuxi, Tang Yaoping. Effect and mechanism of exosome-like vesicles derived from Trichosanthes kirilowii Maxim. in preventing and treating atherosclerosis [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1768-1781. |
| [8] | Liu Hongtao, Wu Xin, Jiang Xinyu, Sha Fei, An Qi, Li Gaobiao. Causal relationship between age-related macular degeneration and deep vein thrombosis: analysis based on genome-wide association study data [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1602-1608. |
| [9] | Peng Zhiwei, Chen Lei, Tong Lei. Luteolin promotes wound healing in diabetic mice: roles and mechanisms [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1398-1406. |
| [10] | Jia Jinwen, Airefate·Ainiwaer, Zhang Juan. Effects of EP300 on autophagy and apoptosis related to allergic rhinitis in rats [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1439-1449. |
| [11] | Zhang Qian, Huang Dongfeng. Weighted gene co-expression network analysis combined with machine learning to screen and validate biomarkers for osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1096-1105. |
| [12] | Liu Yu, Lei Senlin, Zhou Jintao, Liu Hui, Li Xianhui. Mechanisms by which aerobic and resistance exercises improve obesity-related cognitive impairment [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1171-1183. |
| [13] | Wang Zhengye, Liu Wanlin, Zhao Zhenqun. Advance in the mechanisms underlying miRNAs in steroid-induced osteonecrosis of the femoral head [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1207-1214. |
| [14] | Fu Zhenyi, Li Junhao, Zhang Yating, He Yunkai, Liu Junyu, Wei Yunhao, Liu Jiaxin. Schwann cells promote peripheral nerve regeneration: retrospect and prospect [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1236-1246. |
| [15] | Bao Zhuoma, Hou Ziming, Jiang Lu, Li Weiyi, Zhang Zongxing, Liu Daozhong, Yuan Lin. Effect and mechanism by which Pterocarya hupehensis skan total flavonoids regulates the proliferation, migration and apoptosis of fibroblast-like synoviocytes [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(4): 816-823. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||