Chinese Journal of Tissue Engineering Research ›› 2025, Vol. 29 ›› Issue (3): 554-562.doi: 10.12307/2025.134
Previous Articles Next Articles
Ferroptosis and osteoporosis
Yang Cheng1, Li Weimin1, Ran Dongcheng1, Xu Jiamu1, Wu Wangxiang1, Xu Jiafu1, Chen Jingjing1, Jiang Guangfu2, Wang Chunqing3
- 1Clinical Medicine School of Guizhou Medical University, Guiyang 550004, Guizhou Province, China; 2Department of Orthopedics, Guizhou Provincial People’s Hospital, Guiyang 550004, Guizhou Province, China; 3Department of Emergency, Affiliated Hospital, Guizhou Medical University, Guiyang 550004, Guizhou Province, China
-
Online:2025-01-28Published:2024-06-04 -
Contact:Wang Chunqing, MD, Professor, Department of Emergency, Affiliated Hospital, Guizhou Medical University, Guiyang 550004, Guizhou Province, China -
About author:Yang Cheng, Master candidate, Clinical Medicine School of Guizhou Medical University, Guiyang 550004, Guizhou Province, China -
Supported by:National Natural Science Foundation of China, No. 8206060273 (to WCQ)
CLC Number:
Cite this article
Yang Cheng, Li Weimin, Ran Dongcheng, Xu Jiamu, Wu Wangxiang, Xu Jiafu, Chen Jingjing, Jiang Guangfu, Wang Chunqing. Ferroptosis and osteoporosis[J]. Chinese Journal of Tissue Engineering Research, 2025, 29(3): 554-562.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
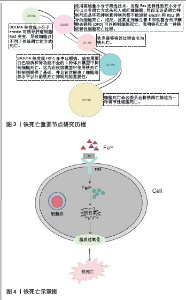
2.1 铁死亡概述 铁死亡的发现经历了漫长的研究过程[21-25], 见图3,而自2012年正式将铁死亡描述为非凋亡性细胞死亡这一术语以来,人们对铁死亡的过程和功能越来越感兴趣[22]。这种由铁依赖性磷脂过氧化驱动的细胞死亡的独特模式受到多种细胞代谢事件的调节,除了与疾病相关的许多信号传导途径相互调控之外,还参与氧化还原稳态、铁代谢、线粒体活性以及氨基酸、脂质和糖的代谢等[26]。 在形态学上,细胞发生铁死亡时,形态表现为细胞较小、较圆,细胞间分离;膜显示完整的细圆细胞膜,质膜出血,无核凝聚;线粒体凹陷,线粒体嵴数量减少或缺失,线粒体膜密度增加,线粒体外膜破裂。在透射电子显微镜下观察到铁死亡的主要形态学特征包括具有退化嵴和膜密度增加的致密且较小的线粒体[27-30]。铁死亡可通过两种主要途径发生,即外源性或转运蛋白依赖性途径和内源性或酶调节途径[31]。含铁酶脂氧合酶是通过产生脂质氢过氧化物而导致铁死亡的主要启动子,其功能依赖于链脂肪酸-CoA连接酶4依赖性脂质生物合成的激活。相反,谷胱甘肽过氧化物酶 4(glu-tathione peroxidase 4,GPX4)目前被认为是铁死亡的中心阻遏物,其活性取决于由胱氨酸-谷氨酸反向转运蛋白SLC 7A 11激活产生的谷胱甘肽。许多代谢(特别是涉及铁、脂质和氨基酸)和降解途径(自噬/自噬和泛素蛋白酶体系统)通过直接或间接调节铁积累或脂质过氧化来协调复杂的铁中毒反应[32]。转录因子NFE 2L2在上调抗铁死亡防御中起着核心作用,而选择性自噬可能促进铁死亡[31]。因此,铁死亡在多个水平上被精确调控,包括表观遗传、转录、转录后和翻译后层面[31]。对于治疗癌症、神经退行性变、组织损伤、炎症和感染,铁死亡广泛参与其生物学过程的调控,在各类疾病的靶点治疗中有着巨大的应用潜力[33]。 2.2 铁死亡的分子机制 铁死亡被定义为一种铁依赖性的并在谷胱甘肽依赖性脂质过氧化修复系统受损时通过活性氧的致死性积累而发生的细胞死亡方式[31],见图4。而铁死亡的关键介质是铁代谢和脂质过氧化[34]。在许多方面,对于铁死亡的研究领域都是起步阶段,近年关于铁死亡已发表的研究论文数量呈指数级增长,因此作者对铁死亡的分子机制做一概述,以讨论新出现的问题和 挑战。 "
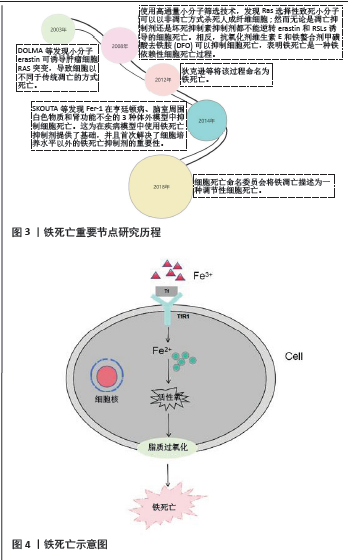

2.2.1 铁代谢 作为生命必需的微量元素之一,铁主要以亚铁离子(Fe 2+)和三价铁离子(Fe 3+)的形式存在于体内。细胞内铁稳态表现为铁的吸收、输出、利用和储存之间的平衡[33]。铁参与能量代谢、酶反应和其他细胞功能[31]。人体的铁调节系统包括全身调节和细胞内调节,系统调节的主要因子是肝分泌产生的铁调节激素,铁调素与铁转运蛋白结合并通过引起其内化而降低其活性,从而抑制铁的吸收和转移;细胞调节依赖于铁调节蛋白/铁反应元件系统。铁调节蛋白参与铁转运和细胞内缓冲系统,并与不稳定铁池中的诱导铁螯合以调节细胞内铁稳态。蛋白质的任何错误翻译都会破坏细胞内的铁代谢,促进细胞毒性和死亡。铁调节蛋白与铁反应元件结合,调节铁代谢中关键蛋白质的合成[35-36]。当然,细胞内铁稳态还可以由其他因素控制,例如缺氧、细胞因子和激素等。铁对于哺乳动物器官系统中细胞的正常功能至关重要,它涉及许多必要的生理活动,包括血红蛋白合成、氧转运、ATP产生和酶促反应[37]。值得注意的是,铁代谢广泛参与骨生长和发育。异常的铁代谢会增加心血管疾病、神经退行性疾病、癌症、骨疾病等的发生率[38]。 在正常情况下,铁代谢受到严格控制,使铁的输入和输出处于动态平衡,以维持机体的生理功能,并且过量的铁会对组织甚至对身体造成损害。当铁死亡发生时,大量游离Fe2+在细胞内聚集。当发生铁死亡时,转铁蛋白携带Fe3+与质膜上的转铁蛋白受体结合。转铁蛋白在质膜上形成小泡,将Fe3+吸附到细胞内,小泡内的低pH值使Fe3+从转铁蛋白上剥离;脱落的Fe3+被还原成Fe2+,在细胞质中解离到细胞内不稳定铁池中或结合到铁蛋白上。最后,铁蛋白可以被NC0A 4介导的自噬溶酶体包裹。该过程称为铁蛋白吞噬,可以降解并释放大量的Fe2+。Fe2+从多不饱和脂肪酸中提取氢原子,产生羟基自由基,可对DNA、蛋白质和膜脂质造成氧化损伤,该过程通过芬顿反应促进脂质过氧化反应的发生,破坏细胞膜并导致细胞死亡。铁蛋白以其惰性形式储存细胞内铁,并且其惰性状态不能促进脂质过氧化。因此,铁蛋白丰度决定了对铁死亡的易感性。当细胞内不稳定铁池变得稀缺时,铁蛋白越多储存的铁就越多,就会抵抗细胞发生铁死亡。相反,铁蛋白耗竭导致铁释放到细胞内不稳定铁池中,最终导致对铁死亡的敏感性增加[31,36]。另一项研究发现,prominin2通过促进含铁蛋白的多泡体和外泌体的形成,将铁携带出细胞,从而有助于抵抗铁死亡[39]。 2.2.2 脂质过氧化 脂质过氧化反应会对生物膜(质膜和内部细胞器膜)、脂蛋白和其他含脂质的分子的造成氧化损伤。多不饱和脂肪酸磷脂是脂质过氧化的重要靶点,脂质过氧化涉及许多疾病和病理状况,例如动脉粥样硬化、缺血再灌注损伤、神经变性疾病和癌症。脂质过氧化是引起铁缺乏症的重要原因,游离的多不饱和脂肪酸是脂质氧化的重要底物,可与磷脂双分子层结合,导致过度氧化,从而引发脂质凋亡。细胞膜中的多不饱和脂肪酸是活性氧攻击的重要靶点。脂质过氧化是一系列自由基的连锁反应。触发脂质过氧化需要脂质双层中多不饱和脂肪酸磷脂的2个碳碳双键之间的二烯丙基氢原子形成碳中心自由基,该自由基与分子氧反应生成过氧自由基,最终导致脂质过氧化[32,40-42]。 氧化还原稳态在维持体内的许多生理、生化和免疫反应中起重要作用。一旦平衡被破坏,则形成氧自由基的链式反应,导致细胞膜完整性的破坏,并最终导致细胞器和细胞膜破裂和功能障碍。氧化应激和脂质过氧化是铁缺乏症的标志。在脂质过氧化过程中,由于活性氧与生物膜相关的多不饱和酸侧链和大分子如核酸之间的脂质过氧化反应,形成大量脂质过氧化物,如丙二醛和4-羟基壬烯醛。脂质过氧化物改变细胞膜的流动性和渗透性,最终导致细胞结构和功能的变化。4-羟基壬烯醛和丙二醛是与DNA碱基、蛋白质和其他亲核分子反应的显著毒性产物,导致严重的细胞毒性。脂质过氧化物检测目前被认为是脂质过氧化水平的最有效指标,丙二醛是最典型的终产物,也是脂质过氧化和铁凋亡观察的反映性指 标[12,43-46]。 尽管科学家们目前对于铁死亡的分子机制有了很大的认识,但由于铁凋亡是一种新的细胞死亡形式,其机制和相关信号通路仍需进一步探索。 2.3 铁死亡对成骨细胞及破骨细胞的效应 骨是一种多功能、高度矿化的结缔组织,作为运动系统的中心,并为所有内脏器官提供结构支撑。值得注意的是,骨骼也是钙和磷的储藏室,这是维持矿物质稳态的先决条件[46]。骨组织代谢特别活跃,在整个生命过程中骨组织都在不断地重塑[47]。人体骨代谢是一个复杂的过程,由成骨细胞诱导的骨形成和破骨细胞介导的骨吸收之间的平衡来维持骨矿物质稳态和强度[48]。当骨骼受到不利因素的影响时,例如衰老、营养不良、酗酒、雌激素减少以及对药物的相关不良反应,骨形成和吸收之间的平衡将被打破,这是产生骨质疏松症的主要原因[49-50]。而自铁死亡这种独特的细胞死亡方式被发现以来,越来越多的研究表明了铁死亡与骨代谢疾病的关系,有研究报道成骨细胞、破骨细胞可以通过不同的途径发生铁死亡,从而参与骨代谢的生物学过程,进而干预骨质疏松症的发生发展。 2.3.1 成骨细胞铁死亡 成骨细胞来源于骨髓中的间充质干细胞,成骨分化在骨骼形成的过程中至关重要,并在骨基质的合成、分泌和矿化中起主导作用[51-52]。成骨细胞可能发生铁死亡,而其发生铁死亡后会影响骨形成的一系列过程[53],已有多项研究报道了两者的作用关系,见表2。"


糖尿病和骨质疏松症是两种常见的疾病,由于糖尿病患者骨质疏松性骨折的发生率显著增加,铁超负荷又会导致骨质流失。为探索三者的关系,LIN等[54]在一项研究中使用高脂高糖食物喂养建立糖尿病大鼠模型,随后在其腹膜内单次注射低剂量链脲佐菌素,发现糖尿病大鼠血清铁蛋白水平(体内铁储存的生物标志物)明显升高,与正常大鼠相比,糖尿病骨质流失大鼠骨组织中SLC7A11和GPX4(铁死亡的抑制性标志蛋白)的表达明显减弱;此外在体外构建的成骨细胞模型中,用高葡萄糖和棕榈酸处理成骨细胞前MC3T3-E1细胞不仅会抑制成骨细胞分化和矿化,还会引发铁死亡相关的成骨细胞死亡。通过进一步的分子机制实验发现,生长激素诱导的成骨细胞铁死亡可能是通过激活METTL3/ASK1-p38信号通路导致糖尿病骨质疏松的主要原因,抑制成骨细胞中的铁死亡可能为糖尿病骨质疏松症提供潜在的治疗策略。另一项研究为进一步探索铁死亡影响成骨分化的具体分子机制,通过柠檬酸铁铵增加细胞内铁含量,导致铁死亡和成骨细胞分化受损[55]。柠檬酸铁铵上调Trfr和DMT1基因的表达以增加铁的摄取,从而积累细胞内不稳定的亚铁以维持铁超载状态;然后,过量的亚铁产生活性氧和脂质过氧化产物,引起铁死亡,并伴有典型的线粒体形态变化,如收缩和凝聚的膜,伴有嵴的缩小和丧失以及外膜破裂;进一步实验数据表明铁死亡是导致柠檬酸铁铵破坏成骨细胞分化的主要原因。从机制上来将,铁剂量依赖性下调Wnt靶基因的表达,抑制Wnt报告基因TopFlash构建体的转录,从而抑制经典的Wnt信号传导。Wnt 激动剂、铁死亡抑制剂或抗氧化剂褪黑激素可逆转铁抑制的典型Wnt 信号转导,通过减少活性氧和脂质过氧化产物的产生来恢复成骨细胞分化,从而预防铁死亡,而不会减少铁过载,且无论是否去除铁过载,都能维持成骨细胞分化。此外,应适当程度使用铁螯合剂,因为铁本身是成骨分化所必需的。XU等[56]探讨了维生素 D 受体激活是否能通过抗铁死亡机制保护机体免受年龄相关性骨质疏松症的影响,维生素 D 受体激活剂1,25(OH)2D3减轻了D-氨基半乳糖诱导成骨细胞的衰老和铁死亡,表现为衰老相关分泌表型基因下调、线粒体形态改善、谷胱甘肽升高以及脂质过氧化标记物减少。体外研究采用了前成骨细胞MC3T3-E1和原代大鼠成骨细胞,1,25(OH)2D3或铁素沉着抑制剂可降低D-氨基半乳糖诱导的成骨细胞的细胞衰老标志物。从机制上讲,1,25(OH)2D3激活了维生素 D 受体及其下游核因子红细胞2相关因子 2(nuclearfactor erythroidderived 2-like 2,Nrf2)/GPX4信号通路,从而导致脂质过氧化的下调。敲除Nrf2或添加GPX4抑制剂(RSL-3)会阻断1,25(OH)2D3对D-氨基半乳糖诱导的铁死亡和衰老的保护作用,维生素 D 受体基因敲除阻碍了1,25(OH)2D3诱导的成骨细胞Nrf2/GPX4通路的激活。蛋白质组学和免疫荧光分析证实,维生素 D 受体基因敲除的小鼠发生了铁死亡和Nrf2/GPX4通路的抑制。最终数据表明,铁死亡在与年龄相关的骨质疏松症中起着至关重要的作用,激活维生素 D 受体可通过刺激Nrf2/GPX4信号通路减轻成骨细胞的铁死亡。JIANG等[57]为了确定铁超载是否会导致成骨细胞铁死亡,并探讨成骨细胞的铁死亡是否参与铁超载诱导的体外和体内骨质疏松症。通过柠檬酸铁铵用于模拟铁超载条件,而去铁胺和铁抑素1用于在体外抑制MC3T3-E1细胞的铁死亡,再利用右旋糖酐铁建立小鼠铁超载模型,进行一系列实验方法对成骨分化功能进行检测后,结果表明铁超载降低了成骨细胞活力、超氧化物歧化酶和谷胱甘肽水平,增加了活性氧的产生、脂质过氧化、丙二醛水平和铁死亡相关蛋白的表达,并诱导了线粒体的超微结构变化,同时在体外抑制成骨分化和矿化,而抑制铁死亡就会逆转上述变化。上述结果表明,成骨细胞的铁死亡在铁超载诱导的骨质疏松症中起着至关重要的作用。 维持铁稳态和靶向成骨细胞的铁死亡可能是治疗或预防铁超载引起的骨质疏松症的潜在措施。有报道称铁死亡参与糖皮质激素引起的骨质疏松症中成骨细胞的分化,为深入阐明其分子机制,确定可能的治疗靶点,LI等[58]采用大剂量地塞米松诱导骨髓间充质干细胞铁死亡,此外,还建立了小鼠类固醇性骨质疏松症模型,然后评估褪黑激素是否可以抑制铁死亡途径,以提供对糖皮质激素诱导的小鼠类固醇性骨质疏松症模型的早期保护,并研究所涉及的信号通路。体外实验证实褪黑激素能显著减轻糖皮质激素诱导的骨髓间充质干细胞铁死亡,通路分析显示褪黑激素通过激活磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶B(proten kinase B,AKT)/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)轴来改善铁死亡。PI3K是铁死亡抵抗的重要调节因子,PI3K激活剂模拟褪黑激素的抗铁死亡作用,但当PI3K通路被阻断时,褪黑激素的作用被减弱。褪黑激素可通过激活PI3K/AKT/mTOR信号通路抑制铁死亡,从而抑制小鼠类固醇性骨质疏松症的发生,因此,褪黑激素有可能成为预防和治疗类固醇性骨质疏松症的新药物。肥胖与骨质疏松症的关系在过去几年中得到了广泛的研究,然而,肥胖对骨骼健康的影响仍然存在争议,其潜在的分子机制尚未完全了解。CHEN等[59]研究表明,高脂饮食诱导的肥胖导致雄性大鼠股骨体积/组织体积、小梁数和皮质厚度显著降低。且肥胖模型大鼠骨组织中铁死亡抑制蛋白SLC7A11和GPX4表达减弱,与血清肿瘤坏死因子α浓度升高相关。铁死亡抑制剂可有效挽救减少的成骨相关H型血管和骨祖细胞,并下调血清肿瘤坏死因子α水平,以改善肥胖大鼠的骨质流失。由于铁死亡和肿瘤坏死因子α都影响骨骼和血管的形成,通过实验进一步研究了铁死亡和肿瘤坏死因子α之间的相互作用,及其对体外成骨和血管生成的影响,结果发现铁死亡抑制剂可以减少肿瘤坏死因子α处理的MG63和脐静脉内皮细胞的细胞内活性氧过量产生,并增强成骨和血管生成。该研究揭示了铁死亡与肿瘤坏死因子α的相互作用及其对成骨和血管生成的影响,为肥胖相关骨质疏松症的发病机制和再生治疗提供了新的见解。 2.3.2 破骨细胞铁死亡 破骨细胞是在巨噬细胞集落刺激因子和核因子κB受体激活因子配体刺激下由造血细胞分化而来的大型多核细胞,具有骨吸收功能[60]。而破骨细胞也可能发生铁死亡,铁离子可通过产生活性氧促进破骨细胞分化和骨吸收。而已有证据表明铁螯合剂在体外可以抑制破骨细胞形成,由此可见,铁死亡对破骨细胞骨吸收的影响也与骨质疏松有重要的联系,见表3。 多项研究表明,干扰素调节因子9不仅调节先天性和适应性免疫反应,而且在炎症、抗病毒反应和细胞发育中发挥重要作用。然而,干扰素调节因子9在破骨细胞中的确切作用尚未见报道。为了阐明干扰素调节因子9在破骨细胞分化中的作用,LAN等[61]建立了绝经后骨质疏松症的卵巢切除小鼠模型,发现干扰素调节因子9在破骨细胞过度活跃的卵巢切除小鼠中表达降低;此外,敲低干扰素调节因子9后增强了体外破骨细胞分化,通过RNA测序发现其敲低富集的差异表达基因与铁死亡有关,且在体外实验中观察到干扰素调节因子9敲低可以通过抑制铁死亡来促进破骨细胞分化,并进一步验证干扰素调节因子9敲低通过激活信号转导和转录激活因子3促进破骨细胞生成来减少铁死亡,这为绝经后骨质疏松症的治疗提供了新方向。另有一项研究证明了破骨细胞在RANKL诱导的分化过程中存在铁死亡,并且它是由铁-饥饿应答反应和铁蛋白吞噬诱导的[62]。从机制上讲,在常氧但非低氧条件下,铁缺乏反应(转铁蛋白受体1增加,铁蛋白减少)后RANKL刺激可诱导铁死亡,这归因于乌头酸酶活性下调。继续对细胞内铁稳态进行研究,发现在常氧条件下,由FTHNCOA 4复合物自噬体降解启动的铁蛋白吞噬过程在RANKL刺激后被激活,有趣的是,这些过程在缺氧条件下无法观察到。此外,该研究还发现缺氧诱导因子1α参与了缺氧条件下铁蛋白吞噬和自噬通量的降低。体内研究表明,缺氧诱导因子1α特异性抑制剂2ME2可预防小鼠卵巢切除骨质疏松模型的骨丢失。总之,该研究全面阐述了体内外破骨细胞铁死亡的作用,并创新性地提出了靶向缺氧诱导因子1α和铁蛋白诱导破骨细胞铁死亡可能成为治疗骨质疏松症的一种新方法。近年来,天然化合物显示出作为绝经后骨质疏松症治疗剂的极大潜力。来自体外破骨细胞生成实验的新证据表明,乌头碱作为破骨细胞分化调节剂而不引起细胞毒性。然而,乌头碱在各种骨质疏松模型中的体内功能需要澄清。为了解决这个问题,XUE等[63]给卵巢切除术诱导的骨质疏松小鼠腹腔注射乌头碱8周,发现可以有效地逆转了卵巢切除小鼠骨质疏松的发展,导致椎骨骨丢失减少和高骨转换标志物的恢复。究其具体的分子机制,乌头碱显著抑制破骨细胞生成,在体内和体外通过减少破骨细胞特异性基因表达,如NFATc 1、c-Fos、组织蛋白酶K和基质金属蛋白酶9的表达;重要的是,乌头碱可以通过抑制GPX4和上调链脂肪酸-CoA连接酶4来调节破骨细胞铁死亡,这是通过抑制核因子κB信号通路中I-kB和p65的磷酸化来实现的。这些结果表明,乌头碱是一种潜在的治疗选择,通过抑制核因子κB信号介导的破骨细胞铁死亡来影响骨质疏松。既往青蒿素化合物被证明通过抑制骨吸收而具有抗骨质疏松症的作用。在破骨细胞分化过程中,破骨细胞通过转铁蛋白受体1介导的转铁蛋白内吞作用吸收大量铁。JIN等[64]的实验表明,转铁蛋白受体1敲低减轻了青蒿琥酯对破骨细胞的细胞毒性作用;此外,抗反转录病毒疗法可有效预防铁超负荷引起的骨质流失。由此可见,青蒿琥酯通过诱导铁死亡来抑制铁摄取刺激的破骨细胞分化,它有可能成为治疗铁超负荷引起的骨质疏松症的潜在药物。QU等[65]探讨了唑来膦酸对破骨细胞生长的抑制作用及其机制,结果表明,F-box蛋白9的表达被唑来膦酸抑制,F-box蛋白9过表达抑制了唑来膦酸调节破骨细胞铁凋亡的作用。F-box蛋白9与p53相互作用,降低了p53蛋白的稳定性。总的来说,唑来膦酸通过触发F-box蛋白9介导的p53泛素化和降解诱导破骨细胞铁死亡影响骨质疏松。在今年最新的一项研究中,ZHANG等[66]发现了一种转录因子——Nrf2,通过表达参与抗氧化过程和铁代谢的基因来调节细胞对氧化应激和铁超载的防御。研究显示Nrf2敲除在正常条件下诱导氧化应激并促进破骨细胞分化,但在铁过载条件下诱导铁死亡;而Nrf2敲除通过抑制破骨细胞分化来缓解铁超载诱导的骨质流失。这些数据都提供了铁死亡与骨质疏松症有关的潜在证据。 2.4 铁死亡在骨质疏松症中的应用 随着对铁死亡研究的不断深入,研究者逐渐开始找寻对其骨质疏松症的治疗靶点,以期对新药物的研发找到突破口,努力实现基础科研到临床治疗的转化。例如:WANG等[67]检测到2型糖尿病骨质疏松大鼠模型骨组织中FtMt的表达和铁死亡的发生;并且发现,FtMt的过表达减少了高糖条件下过量铁诱导的氧化应激,从而抑制成骨细胞中铁死亡的发生,而FtMt的沉默通过活性氧/PINK1(PTEN induced putative kinase 1)/Parkin信号通路诱导2型糖尿病性骨质疏松大鼠模型成骨细胞线粒体自噬,这表明FtMt可能是治疗2型糖尿病性骨质疏松的潜在靶点。 铁螯合剂对铁超载引起的骨质疏松症有很好的治疗作用,目前在临床中使用的3种类型的铁螯合剂是去铁胺、去铁酮和地拉罗司[68]。首先,去铁胺应用于缺乏VHL基因蛋白产物的小鼠中,成骨细胞中的缺氧诱导因子1α活化可显著增加血管分布并产生更多的骨以响应牵张成骨。此外,去铁胺是一种缺氧诱导因子1α激活剂,可促进血管生成和骨生成。成骨细胞中低氧诱导因子途径的条件性激活可增加血管生成和骨生成,并缓解双侧卵巢切除骨质疏松小鼠诱导的骨丢失。但是,尽管对两者的研究不断增加,查阅文献还是可以发现,铁死亡治疗骨质疏松的药物并未在临床上普及,基本上都是停留在动物实验上,故未来还需要研究工作者付出更多的努力,探索出两者治疗相互作用的潜在靶点,早期找到以铁死亡为靶点治疗骨质疏松的新药物。 2.5 总结与展望 随着分子生物学的发展,细胞所存在的多种死亡方式逐渐被科学家们发现。而近年来,铁死亡是细胞众多死亡方式中研究者关注的焦点及难点,其在机体能量代谢紊乱、炎症、氧化应激以及维持机体内环境稳态的平衡中起到重要作用。骨质疏松症依然是国际上临床未解决的重大难题,尽管目前市面上有用于治疗骨质疏松症的药物,但主要是抑制破骨细胞介导的骨吸收,且其疗效往往未尽人意。然而,令人振奋的消息是,铁死亡对骨质疏松症的治疗作用不仅限于抑制骨吸收,除破骨细胞外,成骨细胞、骨细胞和干细胞也受到骨质疏松症中铁死亡调节的影响。通过阐明铁死亡和骨质疏松症的分子机制可以为预防及治疗骨质疏松症提供一个新的突破口,探索出不良反应更少的新疗法来指导骨质疏松症的诊疗。但是铁死亡与骨质疏松症的研究仍处于相对早期的阶段,铁死亡的具体机制、参与的相关信号通路以及调控的分子新靶点尚不清楚。尽管目前对铁死亡在骨质疏松症中的作用仍有许多认识上的空白,但对铁死亡作用于骨质疏松症的信号通路、调控机制和病理意义的深入研究将有助于开发新的骨质疏松症靶向治疗药物。相信在未来的研究中,在科研工作者的努力下,铁死亡与骨质疏松症之间的神秘面纱会被揭开,骨质疏松症的治疗策略将实现质的飞跃。"

| [1] KANIS JA, MELTON LR, CHRISTIANSEN C, et al. The diagnosis of osteoporosis. J Bone Miner Res. 1994;9(8):1137-1141. [2] GIANGREGORIO LM, PAPAIOANNOU A, MACINTYRE NJ, et al. Too Fit To Fracture: exercise recommendations for individuals with osteoporosis or osteoporotic vertebral fracture. Osteoporos Int. 2014;25(3):821-835. [3] LEBOFF MS, GREENSPAN SL, INSOGNA KL, et al. The clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2022; 33(10):2049-2102. [4] 中华医学会骨质疏松和骨矿盐疾病分会. 原发性骨质疏松症诊疗指南(2022)[J]. 中华骨质疏松和骨矿盐疾病杂志,2022,15(6): 573-611. [5] 章振林, 岳华, 李梅, 等. 中国《原发性骨质疏松症诊疗指南(2022版)》要点解读[J]. 诊断学理论与实践,2023,22(3):230-233. [6] COMPSTON JE, MCCLUNG MR, LESLIE WD. Osteoporosis. Lancet. 2019; 393(10169):364-376. [7] REID IR, BILLINGTON EO. Drug therapy for osteoporosis in older adults. Lancet. 2022;399(10329):1080-1092. [8] KENDLER DL, COSMAN F, STAD RK, et al. Denosumab in the Treatment of Osteoporosis: 10 Years Later: A Narrative Review. Adv Ther. 2022; 39(1):58-74. [9] LIANG B, BURLEY G, LIN S, et al. Osteoporosis pathogenesis and treatment: existing and emerging avenues. Cell Mol Biol Lett. 2022; 27(1):72. [10] CREMERS S, DRAKE MT, EBETINO FH, et al. Pharmacology of bisphosphonates. Br J Clin Pharmacol. 2019;85(6):1052-1062. [11] DEEKS ED. Denosumab: A Review in Postmenopausal Osteoporosis. Drugs Aging. 2018;35(2):163-173. [12] LI J, CAO F, YIN HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. [13] ZOU DB, MOU Z, WU W, et al. TRIM33 protects osteoblasts from oxidative stress-induced apoptosis in osteoporosis by inhibiting FOXO3a ubiquitylation and degradation. Aging Cell. 2021;20(7):e13367. [14] YOSHIDA G, KAWABATA T, TAKAMATSU H, et al. Degradation of the NOTCH intracellular domain by elevated autophagy in osteoblasts promotes osteoblast differentiation and alleviates osteoporosis. Autophagy. 2022;18(10):2323-2332. [15] TAO Z, WANG J, WEN K, et al. Pyroptosis in Osteoblasts: A Novel Hypothesis Underlying the Pathogenesis of Osteoporosis. Front Endocrinol (Lausanne). 2020;11:548812. [16] KETELUT-CARNEIRO N, FITZGERALD KA. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J Mol Biol. 2022; 434(4):167378. [17] CHEN L, MIN J, WANG F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378. [18] LIU S, YAO S, YANG H, et al. Autophagy: Regulator of cell death. Cell Death Dis. 2023;14(10):648. [19] YU P, ZHANG X, LIU N, et al. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. [20] WANG X, ZHOU Y, MIN J, et al. Zooming in and out of ferroptosis in human disease. Front Med. 2023;17(2):173-206. [21] DOLMA S, LESSNICK SL, HAHN WC, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285-296. [22] DIXON SJ, LEMBERG KM, LAMPRECHT MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5): 1060-1072. [23] SKOUTA R, DIXON SJ, WANG J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551-4556. [24] GALLUZZI L, VITALE I, AARONSON SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486-541. [25] YANG WS, STOCKWELL BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234-245. [26] JIANG X, STOCKWELL BR, CONRAD M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282. [27] YU H, GUO P, XIE X, et al. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21(4): 648-657. [28] XIE Y, SONG X, SUN X, et al. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem Biophys Res Commun. 2016;473(4):775-780. [29] HAN C, LIU Y, DAI R, et al. Ferroptosis and Its Potential Role in Human Diseases. Front Pharmacol. 2020;11:239. [30] LIANG C, ZHANG X, YANG M, et al. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv Mater. 2019;31(51):e1904197. [31] TANG D, CHEN X, KANG R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107-125. [32] CHEN X, LI J, KANG R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054-2081. [33] GAO Z, CHEN Z, XIONG Z, et al. Ferroptosis - A new target of osteoporosis. Exp Gerontol. 2022;165:111836. [34] SU Y, ZHAO B, ZHOU L, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020;483:127-136. [35] GALY B, CONRAD M, MUCKENTHALER M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2024; 25(2):133-155. [36] WALLACE DF. The Regulation of Iron Absorption and Homeostasis. Clin Biochem Rev. 2016;37(2):51-62. [37] MAO C, LIU X, ZHANG Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586-590. [38] SAWICKI KT, De JESUS A, ARDEHALI H. Iron Metabolism in Cardiovascular Disease: Physiology, Mechanisms, and Therapeutic Targets. Circ Res. 2023;132(3):379-396. [39] BROWN CW, AMANTE JJ, CHHOY P, et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell. 2019; 51(5):575-586. [40] FRIEDMANN AJ, SCHNEIDER M, PRONETH B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180-1191. [41] HAEGGSTROM JZ, FUNK CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111(10): 5866-5898. [42] SHAH R, SHCHEPINOV MS, PRATT DA. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci. 2018;4(3):387-396. [43] LI S, HUANG Y. Ferroptosis: an iron-dependent cell death form linking metabolism, diseases, immune cell and targeted therapy. Clin Transl Oncol. 2022;24(1):1-12. [44] ITO F, SONO Y, ITO T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants (Basel). 2019;8(3):72. [45] LIU P, WANG W, LI Z, et al. Ferroptosis: A New Regulatory Mechanism in Osteoporosis. Oxid Med Cell Longev. 2022;2022:2634431. [46] GAO M, ZHANG Z, SUN J, et al. The roles of circRNA-miRNA-mRNA networks in the development and treatment of osteoporosis. Front Endocrinol (Lausanne). 2022;13:945310. [47] BALLHAUSE T M, JIANG S, BARANOWSKY A, et al. Relevance of Notch Signaling for Bone Metabolism and Regeneration. Int J Mol Sci. 2021; 22(3):1325. [48] MAJIDINIA M, SADEGHPOUR A, YOUSEFI B. The roles of signaling pathways in bone repair and regeneration. J Cell Physiol. 2018;233(4): 2937-2948. [49] LI Y, FENG C, GAO M, et al. MicroRNA-92b-5p modulates melatonin-mediated osteogenic differentiation of bone marrow mesenchymal stem cells by targeting ICAM-1. J Cell Mol Med. 2019;23(9):6140-6153. [50] LI Y, YANG F, GAO M, et al. miR-149-3p Regulates the Switch between Adipogenic and Osteogenic Differentiation of BMSCs by Targeting FTO. Mol Ther Nucleic Acids. 2019;17:590-600. [51] PONZETTI M, RUCCI N. Osteoblast Differentiation and Signaling: Established Concepts and Emerging Topics. Int J Mol Sci. 2021;22(13): 6651. [52] KIM JM, LIN C, STAVRE Z, et al. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells. 2020;9(9):2073. [53] ZHANG H, WANG A, LI G, et al. Osteoporotic bone loss from excess iron accumulation is driven by NOX4-triggered ferroptosis in osteoblasts. Free Radic Biol Med. 2023;198:123-136. [54] LIN Y, SHEN X, KE Y, et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. 2022;36(3):e22147. [55] LUO C, XU W, TANG X, et al. Canonical Wnt signaling works downstream of iron overload to prevent ferroptosis from damaging osteoblast differentiation. Free Radic Biol Med. 2022;188:337-350. [56] XU P, LIN B, DENG X, et al. VDR activation attenuates osteoblastic ferroptosis and senescence by stimulating the Nrf2/GPX4 pathway in age-related osteoporosis. Free Radic Biol Med. 2022;193(Pt 2):720-735. [57] JIANG Z, WANG H, QI G, et al. Iron overload-induced ferroptosis of osteoblasts inhibits osteogenesis and promotes osteoporosis: An in vitro and in vivo study. IUBMB Life. 2022;74(11):1052-1069. [58] LI M, YANG N, HAO L, et al. Melatonin Inhibits the Ferroptosis Pathway in Rat Bone Marrow Mesenchymal Stem Cells by Activating the PI3K/AKT/mTOR Signaling Axis to Attenuate Steroid-Induced Osteoporosis. Oxid Med Cell Longev. 2022;2022:8223737. [59] CHEN X, LIU C, YU R, et al. Interaction between ferroptosis and TNF-alpha: Impact in obesity-related osteoporosis. FASEB J. 2023;37(6): e22947. [60] MOON YJ, ZHANG Z, BANG IH, et al. Sirtuin 6 in preosteoclasts suppresses age- and estrogen deficiency-related bone loss by stabilizing estrogen receptor alpha. Cell Death Differ. 2019;26(11):2358-2370. [61] LAN C, ZHOU X, SHEN X, et al. Suppression of IRF9 Promotes Osteoclast Differentiation by Decreased Ferroptosis via STAT3 Activation. Inflammation. 2024;47(1):99-113. [62] NI S, YUAN Y, QIAN Z, et al. Hypoxia inhibits RANKL-induced ferritinophagy and protects osteoclasts from ferroptosis. Free Radic Biol Med. 2021;169:271-282. [63] XUE C, LUO H, WANG L, et al. Aconine attenuates osteoclast-mediated bone resorption and ferroptosis to improve osteoporosis via inhibiting NF-kappaB signaling. Front Endocrinol (Lausanne). 2023;14:1234563. [64] JIN Y, WU S, ZHANG L, et al. Artesunate inhibits osteoclast differentiation by inducing ferroptosis and prevents iron overload-induced bone loss. Basic Clin Pharmacol Toxicol, 2023;132(2):144-153. [65] QU X, SUN Z, WANG Y, et al. Zoledronic acid promotes osteoclasts ferroptosis by inhibiting FBXO9-mediated p53 ubiquitination and degradation. PeerJ. 2021;9:e12510. [66] ZHANG J, ZHANG L, YAO G, et al. NRF2 is essential for iron-overload stimulated osteoclast differentiation through regulation of redox and iron homeostasis. Cell Biol Toxicol. 2023;39(6):3305-3321. [67] WANG X, MA H, SUN J, et al. Mitochondrial Ferritin Deficiency Promotes Osteoblastic Ferroptosis Via Mitophagy in Type 2 Diabetic Osteoporosis. Biol Trace Elem Res. 2022;200(1):298-307. [68] WU D, WEN X, LIU W, et al. Comparison of the effects of deferasirox, deferoxamine, and combination of deferasirox and deferoxamine on an aplastic anemia mouse model complicated with iron overload. Drug Des Devel Ther. 2018;12:1081-1091. |
| [1] | Ao Xiaojing, Li Kun, Liu Yuhang, Yang Xiaoxuan, Wang Xing, Li Zhijun, Ren Xiaoyan, Zhang Shaojie. Development and application of a three-dimensional digital visualization system for children’s neck acupoints [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1834-1840. |
| [2] | Feng Zhimeng, Sun Ning, Sun Zhaozhong, Li Yuefei, Liu Changzhen, Li Sa. Three-dimensional image reconstruction can safely assist one-hole split endoscope in treatment of #br# L5/S1 far lateral lumbar disc herniation [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1876-1882. |
| [3] | Han Haihui, Ran Lei, Meng Xiaohui, Xin Pengfei, Xiang Zheng, Bian Yanqin, Shi Qi, Xiao Lianbo. Targeting fibroblast growth factor receptor 1 signaling to improve bone destruction in rheumatoid arthritis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1905-1912. |
| [4] | Yu Shuai, Liu Jiawei, Zhu Bin, Pan Tan, Li Xinglong, Sun Guangfeng, Yu Haiyang, Ding Ya, Wang Hongliang. Hot issues and application prospects of small molecule drugs in treatment of osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1913-1922. |
| [5] | Zhao Jiyu, Wang Shaowei. Forkhead box transcription factor O1 signaling pathway in bone metabolism [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1923-1930. |
| [6] | Zhou Jinhai, Li Jiangwei, Wang Xuquan, Zhuang Ying, Zhao Ying, Yang Yuyong, Wang Jiajia, Yang Yang, Zhou Shilian. Three-dimensional finite element analysis of anterior femoral notching during total knee arthroplasty at different bone strengths [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(9): 1775-1782. |
| [7] | Wang Wentao, Hou Zhenyang, Wang Yijun, Xu Yaozeng. Apelin-13 alleviates systemic inflammatory bone loss by inhibiting macrophage M1 polarization [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1548-1555. |
| [8] | Chen Shuai, Jin Jie, Han Huawei, Tian Ningsheng, Li Zhiwei . Causal relationship between circulating inflammatory cytokines and bone mineral density based on two-sample Mendelian randomization [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1556-1564. |
| [9] | Yu Jingbang, Wu Yayun. Regulatory effect of non-coding RNA in pulmonary fibrosis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1659-1666. |
| [10] | Wang Qiuyue, Jin Pan, Pu Rui . Exercise intervention and the role of pyroptosis in osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1667-1675. |
| [11] | Zhu Hanmin, Wang Song, Xiao Wenlin, Zhang Wenjing, Zhou Xi, He Ye, Li Wei, . Mitophagy regulates bone metabolism [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1676-1683. |
| [12] | Yuan Weibo, Liu Chan, Yu Limei. Potential application of liver organoids in liver disease models and transplantation therapy [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1684-1692. |
| [13] | Zhao Jiacheng, Ren Shiqi, Zhu Qin, Liu Jiajia, Zhu Xiang, Yang Yang. Bioinformatics analysis of potential biomarkers for primary osteoporosis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1741-1750. |
| [14] | Hu Taotao, Liu Bing, Chen Cheng, Yin Zongyin, Kan Daohong, Ni Jie, Ye Lingxiao, Zheng Xiangbing, Yan Min, Zou Yong. Human amniotic mesenchymal stem cells overexpressing neuregulin-1 promote skin wound healing in mice [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1343-1349. |
| [15] | Li Dijun, Jiu Jingwei, Liu Haifeng, Yan Lei, Li Songyan, Wang Bin. Three-dimensional gelatin microspheres loaded human umbilical cord mesenchymal stem cells for chronic tendinopathy repair [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1356-1362. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||