Chinese Journal of Tissue Engineering Research ›› 2025, Vol. 29 ›› Issue (19): 4132-4144.doi: 10.12307/2025.066
Previous Articles Next Articles
Mechanism of action and progress of mitophagy, ferroptosis, cuproptosis, and disulfidptosis in Alzheimer’s disease
Liu Dandan1, Qin Hewei1, 2
- 1School of Rehabilitation Medicine, Henan University of Chinese Medicine, Zhengzhou 450046, Henan Province, China; 2Department of Rehabilitation Medicine, Second Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou 450002, Henan Province, China
-
Received:2024-03-04Accepted:2024-04-25Online:2025-07-08Published:2024-09-13 -
Contact:Qin Hewei, PhD, Associate chief physician, School of Rehabilitation Medicine, Henan University of Chinese Medicine, Zhengzhou 450046, Henan Province, China; Department of Rehabilitation Medicine, Second Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou 450002, Henan Province, China -
About author:Liu Dandan, Master, School of Rehabilitation Medicine, Henan University of Chinese Medicine, Zhengzhou 450046, Henan Province, China -
Supported by:Central Plains Youth Top Talent Project Supported by Central Plains Talent Program, No. [2021]44 (to QHW); Henan Province TCM Top Talent Training Project, No. [2021]15 (to QHW); a Special Scientific Research Project on the Establishment of “Double First-class” TCM in Henan Province, No. HSRP-DFCTCM-2023-3-27 (to QHW)
CLC Number:
Cite this article
Liu Dandan, Qin Hewei. Mechanism of action and progress of mitophagy, ferroptosis, cuproptosis, and disulfidptosis in Alzheimer’s disease[J]. Chinese Journal of Tissue Engineering Research, 2025, 29(19): 4132-4144.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
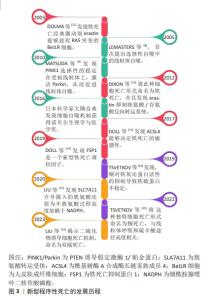
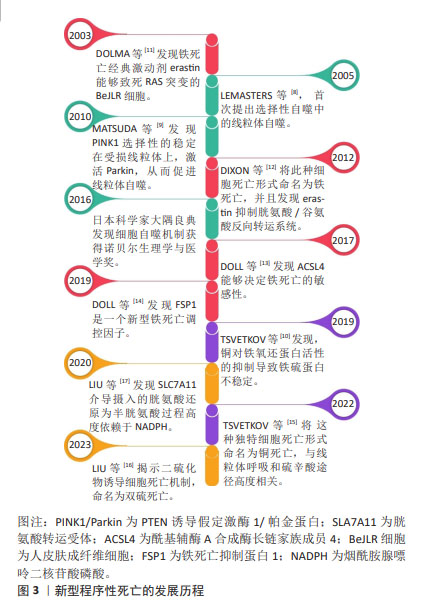
2.1 新型细胞程序性死亡概述 线粒体自噬、铁死亡、铜死亡和双硫死亡是在阿尔茨海默病中逐渐兴起的新型细胞程序性死亡形式,因此接下来主要介绍这4类程序性死亡的发展历程。首先,线粒体自噬作为一种新型的细胞程序性死亡途径,此概念最早由LEMASTERS[8]于2005年正式提出,他观察到线粒体膜电位丧失和线粒体通透性膜孔开放能引起线粒体自噬[8-10]。其次,DOLMA等[11]在2003年首先发现铁死亡经典激动剂erastin能够致死肾素-血管紧张素系统突变的人皮肤成纤维细胞,为发现铁死亡奠定基础。在2012年,DIXON团队[12]发现铁死亡经典途径胱氨酸/谷氨酸反向转运系统,并把这种细胞死亡形式命名为铁死亡。有研究发现酰基辅酶A合成酶长链家族成员4能够通过不饱和脂肪酸途径加重铁死亡的敏感性,并发现新型铁死亡调控因子铁死亡抑制蛋白1[13-14]。随后,TSVETKOV等[15]发现细胞死亡与线粒体呼吸和硫辛酸途径高度相关,并将这种独特细胞死亡形式命名为铜死亡。随后,LIU等[16-17]在2023年揭示由葡萄糖饥饿引起的二硫化物应激诱导细胞死亡机制为双硫死亡。新型程序性死亡的发展历程见图3。"
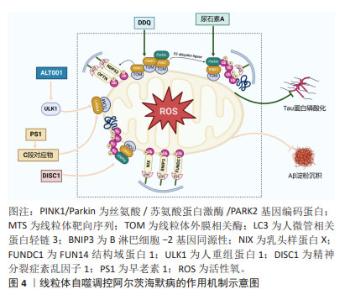
细胞程序性死亡是生物体通过发育程序和应激诱导的信号刺激膜结合蛋白和胞质蛋白,进而诱导复杂的转录变化和翻译后蛋白修饰的一类细胞死亡途径[18]。据报道,在成人身体中,每天有700亿-1 000亿个细胞发生程序性死亡,以维持细胞正常更新和生理稳态[19]。程序性死亡的形态学特征主要有细胞大小改变、细胞膜起泡、细胞质空泡化、DNA缩合以及蛋白质降解等,目前研究新兴的细胞死亡模式主要包括线粒体自噬、铁死亡、铜死亡和双硫死亡等[20]。细胞程序性死亡的异常调节与神经退行性疾病的病理生理过程密切相关,同时这也为探索更多防治细胞程序性死亡紊乱的作用机制提供了新的研究方向[21]。 2.2 线粒体自噬与阿尔茨海默病 2.2.1 线粒体自噬的内在机制 自噬包括选择性和非选择性两种类型,选择性自噬通过清除功能失调的或多余的代谢物质及细胞器,其中线粒体自噬即属于选择性自噬的范畴。线粒体一分为二分裂完成后,自噬系统精准靶向受损线粒体,并与溶酶体结合进行降解,从而清除过度的活性氧以维持线粒体质量控制,此过程称为线粒体自噬[22],其形态学特征表现为胞质无定形,核碎断固缩,形成大量吞噬泡[22]。首先,丝氨酸/苏氨酸蛋白激酶/帕金蛋白信号通路是介导线粒体自噬最经典研究最深入的通路。含有线粒体靶向序列PTEN诱导假定激酶1能够特异地聚集在去极化线粒体上[23]。帕金蛋白是一种E3泛素连接酶,广泛存在于细胞质中[24]。当线粒体受损导致膜电位减少时,PTEN诱导假定激酶1向内膜的输入受到抑制进而聚集在线粒体外膜上[25],聚集在外膜上的PTEN诱导假定激酶1与线粒体外膜相关酶形成复合物,促进PTEN诱导假定激酶1激活帕金蛋白。PTEN诱导假定激酶1首先磷酸泛素化帕金蛋白的N-末端泛素样结构域Ser65位点。由于帕金蛋白锌指结构域RING具有典型的泛素连接酶环状结构,它可以与E2泛素连接酶结合,并将泛素转移到RING中的半胱氨酸(Cys431)中,形成泛素-硫酯中间体。而N-末端泛素样结构域在Ser65位点的磷酸化以及RING结构域的突变会破坏N-末端泛素样结构域与RING的结合,增加帕金蛋白对激活的敏感性,导致帕金蛋白的空间构象发生改变,转化为E3泛素连接酶,进而通过与E2泛素结合酶的协同作用促进多种线粒体蛋白泛素化,使得受损线粒体被磷酸化泛素链包裹[25]。最终,在人微管相关蛋白轻链3 复合物存在的情况下,自噬体吞噬受损线粒体并与溶酶体融合直至降解[26]。同时,PTEN诱导假定激酶1可通过独立于招募帕金的方式触发线粒体自噬。当线粒体功能失调时,位于线粒体外膜上的PINK1可将自噬受体含钙结合结构域和视神经病变诱导反应蛋白招募到线粒体中,随后激活自噬启动子人重组蛋白1诱导线粒体自噬产生[27]。线粒体外膜上有许多可以与LC3结合的受体,这些受体可以绕过PINK1/Parkin途径介导的泛素化过程,直接启动线粒体自噬[28]。LC3受体主要包括B淋巴细胞-2基因同源性、乳头样蛋白X和FUN14结构域蛋白1等。 2.2.2 线粒体自噬在阿尔茨海默病中存在的实验证据 神经元的能量代谢与信号转导需要严重依赖线粒体来支持其功能,因此线粒体自噬通过有效降解受损的神经元内线粒体,从而在神经元健康和功能中起着至关重要的作用。在神经元中,大多数溶酶体所在的体细胞中能够清除自噬底物。线粒体自噬受损主要发生在死后人类阿尔茨海默病脑组织以及阿尔茨海默病诱导多功能干细胞衍生的神经元和阿尔茨海默病动物模型中[29]。研究表明,阿尔茨海默病中的磷酸化的人重组蛋白1与认知正常的对照样本相比出现降低的现象,这说明阿尔茨海默病患者死后脑组织中线粒体自噬机制的启动受损,同时对秀丽隐杆线虫的研究发现,Aβ和Tau诱导的突变体抑制PTEN诱导假定激酶1、乳头样蛋白X或帕金蛋白依赖性线粒体自噬通路[29]。因此,阿尔茨海默病中存在线粒体自噬异常表达。然而,阿尔茨海默病中异常的线粒体自噬与Aβ沉积和Tau磷酸化程度有关。线粒体自噬调控阿尔茨海默病的作用机制见图4。"
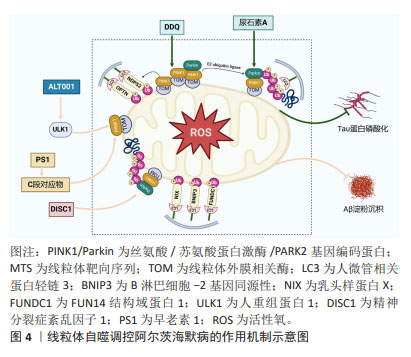

2.2.3 线粒体自噬通过调控Aβ沉积和Tau蛋白磷酸化改善阿尔茨海默病的作用机制 近期PTEN诱导假定激酶1/帕金蛋白介导线粒体自噬靶向调节阿尔茨海默病中Aβ沉积的研究已逐渐兴起。研究显示,进行性Aβ积累显著诱导帕金蛋白募集到去极化的线粒体,并伴有细胞溶质帕金蛋白耗竭[30]。有研究者研究了一种新型小分子线粒体自噬诱导剂ALT001,发现ALT001通过人重组蛋白1依赖性替代线粒体自噬通路诱导线粒体自噬,从而缓解APP突变体诱导的线粒体功能障碍,减轻Aβ蛋白积累,最终逆转PS2APP和5xF阿尔茨海默病小鼠认知障碍缺陷[31]。同时研究显示,早老素1能够调节调节PTEN诱导假定激酶1的表达。其中γ分泌酶催化核心中的早老素1能够导致Aβ及其C端对应物的产生,这种对应物又能反过来上调PTEN诱导假定激酶1和人微管相关蛋白轻链3水平,使其线粒体自噬增强,改善阿尔茨海默病[32]。新型线粒体自噬受体即精神分裂症紊乱因子1已被证明在阿尔茨海默病人脑样本和APP/PS1小鼠中减少[33]。WANG等[33]研究显示,在体外精神分裂症紊乱1的siRNA抑制阻断了Aβ或者自噬激活剂CCCP诱导的线粒体自噬;而过表达精神分裂症紊乱1能够促进PTEN诱导假定激酶1/帕金蛋白通路介导的线粒体自噬,恢复线粒体外膜复合物转移酶或线粒体相关内质网膜的异常表达,减少线粒体中过量的Aβ沉积,进而保护神经突触免受Aβ积累诱导的毒性。 此外,运动对缓解阿尔茨海默病也具有一定作用,ZHAO等[34]研究发现,对APP/PS1小鼠进行运动跑步机训练,小鼠海马体中的沉默调节蛋白1-叉头转录因子1/3轴的激活能够上调PTEN诱导假定激酶1/帕金蛋白通路介导的线粒体自噬活性,减少Aβ蛋白斑块的积累,并改善学习和记忆能力的丧失。线粒体自噬除了能够抑制Aβ沉积外,也与阿尔茨海默病中tau蛋白磷酸化有密切联系。尿石素A作为一种植物化学物质,能够通过启动哺乳动物的线粒体自噬机制,从而对线粒体功能和稳态产生重要影响。研究表明,FANG等[29]发现Tau诱导的APP/PS1小鼠在尿石素A干预下能够激活PTEN诱导假定激酶1/帕金蛋白通路依赖性线粒体自噬,进而减少Tau磷酸化导致认知功能障碍和病理改变。同时,在3xTg阿尔茨海默病小鼠中,给予尿石素A治疗也能改善小鼠记忆障碍并显著降低Tau过度磷酸化[35]。目前已有相关分子能够通过靶向线粒体自噬进而调控阿尔茨海默病中的Tau蛋白磷酸化,减少疾病发生。例如,PRADEEPKIRAN等[36]最近开发的一种分子DDQ已被证明可以减少异常的蛋白质-蛋白质相互作用,并保护神经元免受疾病过程中突变蛋白诱导的毒性。DDQ通过增强沉默调节因子3活性并激活小鼠海马神经元细胞系中PTEN诱导假定激酶1介导线粒体自噬通路,使得p-tau蛋白和总tau蛋白含量显著降低,最终减轻阿尔茨海默病病理。除此之外,自噬受体(如视神经病变诱导反应蛋白)能够以帕金非依赖性方式激活线粒体自噬进而抑制APP/PS1小鼠的记忆丧失[37]。"

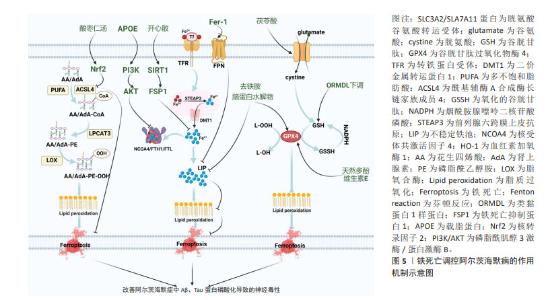
2.3 铁死亡与阿尔茨海默病 铁死亡是一种新型程序性细胞死亡形式,通常以胱氨酸/谷氨酸转运体抑制、铁代谢异常和脂质代谢增多等为特点[38],其形态学特征主要存在于线粒体中,包括细胞核完整但小线粒体增多、线粒体膜密度增加、线粒体嵴破裂或消失以及线粒体外膜破裂[39]。铁死亡调控阿尔茨海默病的作用机制如图5所示。"
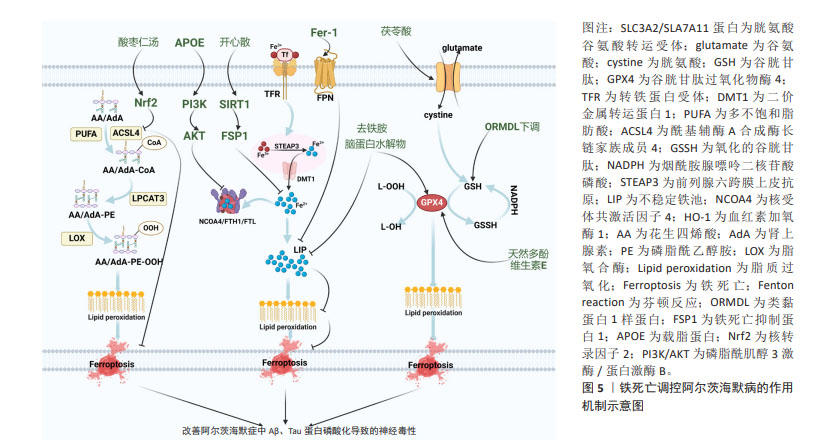

2.3.1 基于胱氨酸/谷氨酸的铁死亡途径调控阿尔茨海默病 systemXc-是由轻链溶质载体族7成员11(solute carrier family 7 member 11,SLC7A11)和重链溶质载体族3成员2组成一种异质二聚体氨基酸转运蛋白,SLC7A11负责将细胞外的胱氨酸转运入细胞内[40],随后转运入胞内的胱氨酸被还原为半胱氨酸进而参与谷胱甘肽的合成[41]。谷胱甘肽会在谷胱甘肽过氧化物酶4作用下降解[41]。当胱氨酸/谷氨酸转运体被抑制时,谷胱甘肽合成减少,导致具有抑制磷脂氢过氧化物合成的谷胱甘肽过氧化物酶4减少,进而引发铁死亡[42]。同样,p53作为一种抑癌基因,能够抑制谷胱甘肽过氧化物酶4系统中的SLC7A11,影响胱氨酸的转运,加重铁死亡[43]。铁死亡是一种非凋亡的细胞死亡形式,谷胱甘肽依赖性的谷胱甘肽过氧化物酶4是抑制铁死亡的中枢因子,同时也是催化哺乳动物细胞中磷脂氢过氧化物还原的主要酶[44]。谷胱甘肽过氧化物酶4可作为谷胱甘肽的调节因子,通过调控谷胱甘肽-二硫化物还原酶,抑制NADPH/H+向谷胱甘肽提供电子,进而将谷胱甘肽转变为氧化型谷胱甘肽。谷胱甘肽过氧化物酶4还能催化毒性脂质氢过氧化的过氧键转变为羟基,失去过氧化物活性,形成相应的醇[45]。相反,氧化的谷胱甘肽能在烟酰胺腺嘌呤二核苷酸磷酸作用下还原为谷胱甘肽[45]。 近年来大量研究表明,铁死亡作为一种新型的铁依赖性程序性细胞死亡方式,能够造成阿尔茨海默病中神经细胞的死亡。脂质过氧化是铁死亡的驱动力,与阿尔茨海默病大脑的神经变性有关,谷胱甘肽过氧化物酶4是海马和皮质神经元中膜脂质氢过氧化物的关键酶,研究报道显示,在谷胱甘肽过氧化物酶4敲除的阿尔茨海默病小鼠模型中,他莫昔芬干预导致谷胱甘肽过氧化物酶4消融表现出海马和皮质神经元蛋白降低、脂质过氧化物增加,但使用抗铁死亡活性的脂溶性抗氧化剂维生素E后逆转了上述变化,改善由铁死亡诱导的神经变性和认知损伤,减轻阿尔茨海默病突触损失及神经元功能障碍[46]。此研究也表明阿尔茨海默病中的确有铁死亡的存在。与此同时,CHEN等[47]研究发现,曾与抗氧化应激有关的脑蛋白水解物也能够通过抑制铁死亡来治疗阿尔茨海默病,在APP/PS1双转基因小鼠中,与模型组相比,脑蛋白水解物能够降低谷胱甘肽过氧化物酶4和血浆铁水平,限制阿尔茨海默病病理标记物Aβ蛋白积累并下调p-Tau/Tau比值,进而减轻神经元损伤,改善阿尔茨海默病小鼠学习记忆功能。有研究表明,外周血血清类黏蛋白1样蛋白3作为铁死亡调控阿尔茨海默病的重要调节因子,其过表达能够抑制谷胱甘肽过氧化物酶4和谷胱甘肽水平,增加丙二醛,引发铁死亡,加重阿尔茨海默病的认知障碍[48]。这对于靶向下调类黏蛋白1样蛋白3来治疗阿尔茨海默病发挥重要的潜在价值。除此之外,中药及中医传统技术对靶向胱氨酸/谷氨酸的介导的铁死亡治疗阿尔茨海默病具有积极作用。有研究发现,天然存在的多酚作为双效治疗剂,其中单个多功能分子单宁酸结合在谷胱甘肽过氧化物酶4的激活剂位点,上调铁死亡抑制因子,减轻淀粉样蛋白和tau蛋白级联反应,为通过激活谷胱甘肽过氧化物酶4治疗阿尔茨海默病提供了一种创新性方法[49]。有研究发现,在APP/PS1小鼠模型中,茯苓酸能够激活核转录因子2/SLC7A11/谷胱甘肽过氧化物酶4信号通路,进而增强神经元的抗氧化功能,增加谷胱甘肽过氧化物酶4和谷胱甘肽含量,减少铁过度积累,从而抑制神经元铁死亡,改善阿尔茨海默病小鼠的认知功能[50]。 2.3.2 基于铁代谢的铁死亡途径调控阿尔茨海默病 铁的摄取、储存和利用异常是细胞内导致脂质过氧化物积累和执行铁死亡所必需的[51]。细胞中铁的摄取是指胞外的Fe3+通过细胞膜上的转铁蛋白转至细胞内的过程[52]。内体囊泡酸化触发Fe3+通过前列腺六跨膜上皮抗原3还原为Fe2+,然后Fe2+被二价金属转运体1释放到细胞质中的不稳定铁池中,最终,过量Fe2+通过芬顿反应产生活性氧引发铁死亡[52]。多余的Fe2+可储存在铁蛋白重链和轻链中,当机体需要额外铁时,Fe2+可通过细胞膜上的铁转运蛋白输出膜外[53]。此外,核受体共激活因子4与铁蛋白的结合增多,可引发自噬体形成,释放更多Fe2+[54]。血红素加氧酶1是血红素降解途径中的限速酶,可释放血红蛋白中的Fe2+,增加细胞内游离铁的积聚[55]。 铁死亡调节功能的紊乱被认为是造成阿尔茨海默病的新型程序性死亡之一。LI等[56]研究发现,在高铁膳食诱导的APP/PS1小鼠阿尔茨海默病模型中,脑组织中转铁蛋白降低,二价金属转运体1表达升高,促进游离亚铁离子向细胞质中输出,诱导铁的再分布,导致Aβ斑块形成和神经元变性,加剧阿尔茨海默病的发生发展。AYTON等[57]在阿尔茨海默病患者死后脑组织中发现,颞下叶铁负荷升高可能通过诱导铁死亡来增加神经元缠结和Aβ斑块形成,进而导致认知功能下降,验证了铁死亡与阿尔茨海默病发病机制的相关性。APOE基因的等位基因变异为散发性阿尔茨海默病带来了最大的遗传风险,有研究证明高丰度的 APOE 能够通过激活磷脂酰肌醇3激酶/蛋白激酶B通路,进而抑制铁自噬蛋白核受体共激活因子4,避免铁依赖性脂质过氧化,最终抑制铁死亡,改善阿尔茨海默病的发生[58]。同时,铁稳态紊乱会加剧阿尔茨海默病病理变化的氧化损伤和认知缺陷。 去铁胺是一种铁螯合剂,可降低铁中毒引起的疾病,ZHU等[59]基于麦芽糖酸铝在阿尔茨海默病大鼠中引起的学习记忆能力障碍,使用铁螯合剂去铁胺后,线粒体膜密度减少嵴增加,谷胱甘肽过氧化物酶4表达上调,丙二醛和活性氧表达下调,减轻铁超载介导的氧化损伤和认知缺陷,进一步证实铁代谢紊乱在阿尔茨海默病发病机制中的重要作用,表明铁螯合剂去铁胺在治疗铁死亡相关的阿尔茨海默病过程中发挥了重要作用。此外,铁转运蛋白是唯一确定的哺乳动物非血红素铁输出因子,在APPswe/PS1dE9阿尔茨海默病小鼠模型中表达下调,使得铁沉积过度,然而加入铁死亡特异性抑制剂后,有效减少铁代谢紊乱,改善Aβ聚集诱导的神经元死亡和记忆障碍[60]。此外,部分中药在抑制阿尔茨海默病细胞铁代谢介导铁死亡的过程中发挥重要作用。研究报道显示,中药复方开心散是治疗神经退行性疾病的潜在药物,在APP/PS1小鼠阿尔茨海默病模型中,开心散能够上调铁死亡抑制蛋白1,并能激活沉默调节因子1/铁死亡抑制蛋白1信号通路,进而减轻铁积累,抑制神经元铁死亡,显著改善阿尔茨海默病小鼠的学习和记忆缺陷[61]。 2.3.3 基于多不饱和脂肪酸的铁死亡途径调控阿尔茨海默病 花生四烯酸和肾上腺素是最为常见的脂质过氧化物。首先,多不饱和脂肪酸通过酰基辅酶A合成酶长链家族成员4催化花生四烯酸/肾上腺素生成花生四烯酸/肾上腺素-CoA衍生物,然后花生四烯酸/肾上腺素-CoA衍生物和磷脂酰乙醇胺在人溶血卵磷脂酰基转移酶3酯化作用下合成花生四烯酸/肾上腺素-CoA衍生物-磷脂酰乙醇胺复合物,脂氧合酶又把此复合物氧化成相应醇,最终导致脂质过氧化,诱发铁死亡[62]。 脂质过氧化物是激活铁死亡的触发因素,由于脂质过氧化物加速了膜脂的氧化,破坏了质膜的脂质双层结构,从而导致铁死亡的发生[63]。ZHANG等[64]研究表明,Aβ诱导的鼠嗜铬细胞瘤细胞系能够引起铁死亡相关的细胞内脂质代谢变化,加入远志皂苷元之后,酰基辅酶A合成酶长链家族成员4表达下调,谷胱甘肽过氧化物酶4表达上调,进而使得活性氧表达降低,减轻Aβ诱导的氧化应激和脂质代谢紊乱,为阿尔茨海默病的遏制和治疗提供新的思路。线粒体基质中的称为共济蛋白的特殊蛋白质是铁死亡的关键调节因子。WANG等[65]在谷氨酸损伤的神经母细胞瘤细胞中,过表达的共济蛋白使得酰基辅酶A合成酶长链家族成员4水平降低,铁离子浓度降低,同时线粒体膜电位恢复,线粒体形态明显改善,最终抑制铁死亡和线粒体障碍,改善阿尔茨海默病等神经退行性疾病。同时,王梦冉[66]研究表明,在Aβ诱导的阿尔茨海默病细胞模型中,跨膜蛋白16F 能够降低酰基辅酶A合成酶长链家族成员4以及活性氧水平,减轻不饱和脂肪酸衍生物,进而减少脂质过氧化物形成,最终抑制铁死亡介导阿尔茨海默病神经元的损伤,因此该研究提示跨膜蛋白16F可能成为阿尔茨海默病治疗的新靶点。此外,中药汤剂酸枣仁汤已被广泛证明对失眠、抑郁和阿尔茨海默病有积极的治疗作用,LONG等[67]在APP/PS1阿尔茨海默病小鼠中证实,酸枣仁汤能够通过激活核转录因子2信号通路,下调酰基辅酶A合成酶长链家族成员4蛋白表达,进而减轻铁蓄积和脂质过氧化,显著缓解阿尔茨海默病小鼠的学习记忆障碍。然而,目前在多不饱和脂肪酸介导的铁死亡调控阿尔茨海默病的研究还有具有较大的研究空间,未来可通过靶向酰基辅酶A合成酶长链家族成员4来探究铁死亡,为阿尔茨海默病的治疗和预防提供新视角。 "


2.4 铜死亡和阿尔茨海默病 铜代谢稳态的破坏会导致铜在细胞内过度积累,最终导致细胞死亡[68]。最近TSVETKOV等[15]研究揭示了铜生物学的一个新方面即铜死亡,是由铜引发、依赖于线粒体呼吸的新型细胞死亡途径。细胞内异常积累的铜离子与三羧酸循环中硫辛酸酯化组分直接结合,导致线粒体硫辛酸酯化蛋白寡聚化以及随后出现铁硫簇蛋白的丢失,进而诱发蛋白质毒性应激,导致细胞死亡[15]。三羧酸循环的蛋白质硫辛酸酯化是一种重要的赖氨酸翻译后修饰,它通过酰胺键将硫辛酸酯与蛋白质赖氨酸残基连接起来,对调控三羧酸循环的关键酶非常重要[69]。 目前研究发现铜死亡的内在作用机制包括以下几点:第一,铜结合硫辛酸酰化蛋白并引起寡聚化可以阻断三羧酸循环。铁氧还蛋白1是蛋白质酰化的上游调节因子,而硫辛酸合成酶能够催化硫辛酸生物合成及协助铁氧还蛋白1发挥作用。铁氧还蛋白1将Cu2+还原为Cu+,并且与硫辛酸合成酶结合促进二氢脂肪酰胺脱氢酶发生硫辛酸酰化,发生硫辛酸酰化后的二氢脂肪酰胺脱氢酶在过量Cu+作用下进行寡聚化,从而抑制线粒体三羧酸循环,促进铜死亡发生[15]。第二,铜引发铁硫蛋白簇丢失,影响线粒体呼吸链,抑制有氧呼吸,引发细胞铜死亡。由于铁硫簇蛋白对细胞内氧化还原变化特别敏感,线粒体内活性氧增多能够导致铁硫簇蛋白当中的[4Fe-4S]和[2Fe-2S]发生互换,进而致使铁硫簇蛋白功能紊乱,加重铜死亡[70]。 第三,谷胱甘肽是重要的内源性铜螯合剂。谷胱甘肽能够将Cu+进行螯合,进而清除细胞内存在的过量Cu+。因此,铁死亡调控通路中的Xc-转运体可能也影响铜死亡调控,包括调控胱氨酸摄取或者谷氨酰胺代谢等,最终影响谷胱甘肽对铜的螯合作用[71]。第四,铜离子载体伊利司莫可以直接将Cu2+载入细胞,并被铁氧还蛋白1还原为Cu+,进而诱导细胞铜死亡的发生[72]。第五,p53可能通过多途径对铜死亡调控进行双向调节。p53通过对谷胱甘肽生成的双重调控作用影响铜死亡,p53作用于细胞周期蛋白依赖性激酶抑制剂能够促进谷胱甘肽合成,进而增加螯合铜的作用,抑制铜死亡,但p53还能够抑制SLC7A11转运体功能,随之抑制谷胱甘肽合成,加重铜死亡[73];p53促进谷氨酰胺酶2功能,调控谷氨酰胺酶代谢,增加谷氨酸水平,进而合成谷胱甘肽[74];p53也可以直接参与铁硫蛋白簇生物合成,增强线粒体代谢,进而抑制铜死亡的进一步加重[75]。 铜稳态失衡是脑损伤的关键机制之一。在阿尔茨海默病中,铜可以通过芬顿反应产生反应性自由基,并直接与Aβ斑块和淀粉样蛋白前体蛋白相互作用,从而促进Aβ斑块的合成和聚集[76]。铜还可以直接与Aβ斑块结合,促进活性氧产生[76]。同时,有研究证实,从Aβ肽中螯合铜可以阻碍其积累并最大限度地提高Aβ降解抑制羟基自由基的产生和氧化损伤,并最终减少细胞死亡[77]。据报道,在暴露于Cu环境下的人神经母细胞瘤细胞通过其5’非翻译区的mRNA增强APP翻译及Aβ淀粉样变性,同时提高铜暴露环境中的APP/PS1 阿尔茨海默病小鼠中相关促炎细胞因子的水平[78]。上述研究表明,阿尔茨海默病的发病机制受铜代谢的影响,而过量Cu+积聚能够引发细胞铜死亡。有研究在C57BL/6J小鼠和海马神经元暴露于高铜环境中发现,过量摄入铜不仅增加小鼠肾脏的Cu排泄量,更促进小鼠海马体内Cu的积累。具体机制表现为铜转运蛋白1表达上调,促进Cu+胞内积累及二氢脂肪酰胺脱氢酶寡聚化,进而导致铁硫簇缺失和热休克蛋白70的表达增加,同时行为学检测中小鼠平台潜伏期和目标区域逃逸延迟,诱导小鼠认知障碍的产生。此外,用铜螯合剂BCS去除Cu后,小鼠海马神经元细胞中细胞凋亡水平和上述铜死亡相关蛋白表达的变化部分恢复,表明铜引起的神经元细胞死亡可能是由铜死亡作用介导的[79]。此外,LAI等[80]使用310个阿尔茨海默病临床样本、加权相关网络分析算法和机器学习工具鉴定了2个与铜死亡相关的分子基因簇,这些基因簇使得免疫应答和免疫浸润升高,与Aβ水平和β分泌酶活性高度相关。此外,使用生物信息学方法,分别从基因数据库中鉴定出7个与阿尔茨海默病相关的铜死亡中心基因和6个靶向这些基因的药物[81]。同时陈桂林等[82]通过生物信息学探讨阿尔茨海默病患者NK细胞与铜死亡机制的相关性,并通过实验验证铁氧还蛋白1、ATP 酶铜转运α、丙酮酸脱氢酶和二氢硫辛酰胺琥珀酰转移酶基因在阿尔茨海默病中存在差异性表达,这为阿尔茨海默病的诊疗提供了潜在的研究思路和靶点。 铜死亡调控阿尔茨海默病的作用机制总结见表1。"


"


2.5 双硫死亡与阿尔茨海默病 双硫死亡是一种新型细胞死亡类型,这种细胞死亡是过量胱氨酸积累引起的二硫化物应激导致的,它不能被常规的细胞死亡抑制剂所抑制,也无法通过敲除铁死亡/细胞凋亡的关键基因来阻止[83]。如前所述,SLC7A11也是铁死亡及铜死亡关键调节因子之一,因此被认为是靶向铁死亡和铜死亡的有效靶标。正常情况下,胱氨酸谷氨酸转运受体系统中的SLC7A11转运体将胱氨酸转运入细胞。当细胞内胱氨酸谷氨酸转运受体充足时,过量的胱氨酸将转化为半胱氨酸,最终半胱氨酸生成谷胱甘肽进而还原活性氧,降低脂质过氧化物表达,维持细胞正常的生理稳态[84]。而胱氨酸谷氨酸转运受体含量的影响因素涉及磷酸戊糖途径,即葡萄糖以的6-磷酸葡萄糖为起始物在6-磷酸葡萄糖脱氢酶催化下形成6-磷酸葡萄糖酸,进而生成5-磷酸核糖及催化NADP+转换为烟酰胺腺嘌呤二核苷酸磷酸[85]。在葡萄糖饥饿或使用葡萄糖转运体抑制剂阻碍磷酸戊糖途径后,烟酰胺腺嘌呤二核苷酸磷酸生成减少,胱氨酸转化为半胱氨酸受阻。过量胱氨酸聚集于细胞内,触发细胞中二硫键应激,导致肌动蛋白细胞骨架之间形成二硫键,F-肌动蛋白收缩并脱离质膜,最终导致细胞发生双硫死亡[16]。同时SLC7A11的高表达导致胱氨酸摄入过量也有助于烟酰胺腺嘌呤二核苷酸磷酸的消耗,导致双硫死亡发生[16]。 肌动蛋白细胞骨架是阿尔茨海默病依赖性突触可塑性中的关键元件,它能够参与调节突触传递机制的不同方面。首先,肌动蛋白丝能够重塑树突棘以响应接收到的刺激。其次,肌动蛋白聚合是网格蛋白介导的内吞作用的驱动力并参与囊泡分选,从而控制谷氨酸受体水平及淀粉样蛋白级联反应,影响Aβ产生[86]。随着二硫键形成肌动蛋白细胞骨架蛋白迁移中断,F-肌动蛋白收缩,从细胞的质膜上分离,最终导致双硫死亡[16]。因此,二硫键的积累和双硫死亡的机制能够破坏肌动蛋白细胞骨架,表明双硫死亡与阿尔茨海默病之间存在潜在联系。随着双硫死亡的潜在机制被发现,双硫死亡相关基因与阿尔茨海默病之间的关系仍不清楚。ZHU等[87]利用基因表达综合数据库分析发现正常样本和阿尔茨海默病样本之间共有8个差异表达基因,其中SLC7A11和人糖原合成酶1在阿尔茨海默病样本中表达上调,随后,CIBERSORT算法显示阿尔茨海默病和对照样本之间的免疫细胞存在显着差异。这些结果初步表明阿尔茨海默病患者中可能存在双硫死亡,同时双硫死亡相关簇还在阿尔茨海默病免疫细胞中发挥重要作用[87]。研究人员发现SLC7A11高表达在葡萄糖饥饿下会造成新的细胞死亡类型,抑制二硫键积累可防止这种死亡,表明二硫键分子的细胞内脂生成是SLC7A11高表达引起死亡的必要条件[16],因此未来有必要通过靶向抑制二硫键分子来探究治疗阿尔茨海默病的潜在作用靶点。此外,葡萄糖转运体抑制剂BAY-876能有效抑制肾癌细胞的葡萄糖摄取,导致烟酰胺腺嘌呤二核苷酸磷酸的产生减少,进而使肌动蛋白细胞骨架蛋白二硫键的异常形成和F-肌动蛋白网络的崩溃,最终促进二硫应激的发生[17]。故在阿尔茨海默病研究当中可以以葡萄糖转运体抑制剂为突破口深入研究在双硫死亡内在机制中与阿尔茨海默病相关的Aβ样蛋白及tau磷酸化的关系。然而,目前关于双硫死亡在阿尔茨海默病中的病理机制研究处于生物信息学分析阶段,基础及临床研究还有待于探索。双硫死亡调控阿尔茨海默病的作用机制总结见表2。"

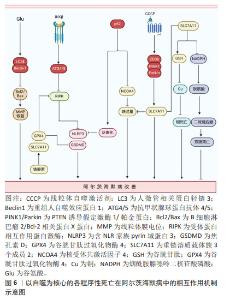
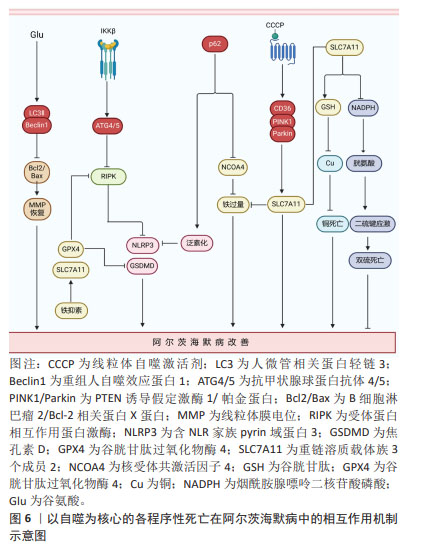
2.6 以自噬为核心的各程序性死亡在阿尔茨海默病中的相互作用 在阿尔茨海默病进展的全过程中,程序性死亡的多模式结合方式均可发挥重要作用。自噬在阿尔茨海默病中作为串扰各细胞程序性死亡的重要关键节点发挥着重要作用。自噬是将自身内部的蛋白或细胞器吞噬包裹形成囊泡,随后与溶酶体融合,导致内容物分解和自噬底物回收进而引起自我消化的细胞程序死亡形式[88]。自噬在阿尔茨海默病中能够分别串扰凋亡、坏死性凋亡、焦亡、铁死亡及铜死亡等细胞程序性死亡形式。 2.6.1 阿尔茨海默病中自噬能够调控凋亡的发生 XIE等[89]在谷氨酸诱导的阿尔茨海默病细胞模型中发现谷氨酸刺激能够显著提高自噬因子重组人自噬效应蛋白1和人微管相关蛋白轻链3蛋白表达,并降低B细胞淋巴瘤2/Bcl-2相关蛋白X蛋白比值及p62蛋白表达,但这些结果在施用自噬抑制剂3-MA处理后,半胱氨酸蛋白酶3表达升高,B细胞淋巴瘤2/Bcl-2相关蛋白X蛋白比值降低,线粒体膜电位得到恢复,减轻凋亡的发生,为治疗阿尔茨海默病提供了新思路。 2.6.2 阿尔茨海默病中以自噬与坏死性凋亡为核心的串扰可能引发焦亡和铁死亡 有研究发现,在Aβ诱导的神经母细胞瘤细胞中,IKKβ激酶激活可以增强自噬,当沉默自噬相关因子抗甲状腺球蛋白抗体4/5时,显著增加了细胞中坏死性凋亡效应因子磷酸化受体蛋白相互作用蛋白激酶1表达,加剧坏死性凋亡的发生,表明在Aβ诱导的神经母细胞瘤细胞中自噬可抑制RIPK诱导的坏死性凋亡,进一步研究发现,自噬上游信号分子IKKβ激酶激活可抑制自噬受损介导的坏死性凋亡,减少Aβ的沉积,对改善阿尔茨海默病起一定治疗作用[90]。混合系激酶域样蛋白是坏死性凋亡的关键执行者,能够通过诱导钾离子外排促进含NLR家族pyrin域蛋白3炎症小体激活,进而引发焦亡的发生,这预示着坏死性凋亡和焦亡之间存在相互作用关系[91]。更有证据表明,在流感病毒感染期间通过激活坏死性凋亡经典通路轴触发含NLR家族pyrin域蛋白3炎症小体激活及随后的等炎症因子释放[91]。此外,坏死性凋亡与铁死亡之间在阿尔茨海默病中也存在一定串扰关系。以往研究发现,在Aβ诱导的阿尔茨海默病大鼠的内嗅皮质中坏死性凋亡可在铁死亡抑制剂铁抑素的阻断作用下,导致铁死亡关键因子谷胱甘肽过氧化物酶4和SLC7A11的表达升高,转铁蛋白受体水平降低,导致受体蛋白相互作用蛋白激酶1/3和混合系激酶域样蛋白表达下降,进而改善坏死性凋亡介导的阿尔茨海默病病理进程。深入研究显示,坏死性凋亡的抑制剂Necrostain-1对铁死亡没有影响,但Aβ诱导的坏死性凋亡被铁抑素抑制,表明铁死亡途径可作为坏死性凋亡的上游信号共同调控阿尔茨海默病中的Aβ神经毒性[38]。 2.6.3 阿尔茨海默病中以自噬与焦亡为核心的串扰可能影响铁死亡 在5XFAD/含NLR家族pyrin域蛋白3敲除小鼠模型中,研究人员发现p62能使小胶质细胞中的促炎反应降低,Aβ斑块沉积减少,认知功能改善。具体机制为P62识别泛素修饰的含NLR家族pyrin域蛋白3并与之结合,进而参与ALP相关的含NLR家族pyrin域蛋白3蛋白降解,最终维持吞噬功能并减轻小胶质细胞的促炎和焦亡状态来改善阿尔茨海默病病理进程[92]。然而,细胞焦亡和铁死亡之间也存在串扰。例如,在骨髓系细胞中,焦孔素D和半胱氨酸蛋白酶11的表达以谷胱甘肽过氧化物酶4敲除程度依赖性方式促进对多种微生物感染的易感性[93]。 2.6.4 阿尔茨海默病中以自噬与铁死亡为核心的串扰可能影响铁死亡和双硫死亡 据报道,自噬在铁稳态的调节中起着重要作用。过量铁能与铁蛋白相互作用并与核受体共激活因子4结合诱发选择性自噬,释放更多游离铁。同时,p62可以导致铁蛋白自噬异常表达,从而诱发铁代谢积累和铁过载[94]。同时,阿尔茨海默病中线粒体自噬和铁死亡之间也存在串扰。LI等[95]通过体内实验建立Aβ诱导的阿尔茨海默病损伤细胞模型,发现CD36/PTEN诱导假定激酶1/帕金蛋白信号通路介导线粒体自噬和铁死亡串扰,而这种铁死亡途径可被自噬激动剂CCCP改善,但被线粒体自噬抑制剂Mdivi-1加剧,这表明此通路引起线粒体自噬在调控铁死亡和抑制阿尔茨海默病淀粉样蛋白的沉积方面发挥重要作用。因此,自噬系统的失调可能直接导致阿尔茨海默病大脑细胞中的铁积累,进而引发铁死亡。而铁死亡抑制因子SLC7A11与铜死亡及双硫死亡密切相关。SLC7A11表达升高,能够促进谷胱甘肽合成,降低活性氧,抑制脂质过氧化生成进而防止铁死亡的发生;SLC7A11水平上调,促进半胱氨酸合成谷胱甘肽,进而螯合铜离子,降低细胞内铜离子水平,抑制铜死亡发生[71];相反,高表达的SLC7A11能够消耗大量烟酰胺腺嘌呤二核苷酸磷酸,使得胱氨酸转变为半胱氨酸受阻,进而触发细胞内二硫键积累,引发二硫死亡[16]。虽然目前阿尔茨海默病在铜死亡和双硫死亡中报道不多,但以SLC7A11为枢纽把铁死亡、铜死亡和双硫死亡把联系起来能更好的为日后研究者探索阿尔茨海默病的发病机制提供线索。以自噬为核心的各程序性死亡在阿尔茨海默病中的相互作用,见图6。"

"


2.7 新型程序性死亡在阿尔茨海默病中的临床转化 铁死亡作为一种新型的程序性细胞死亡,已被证明参与阿尔茨海默病的发病机制,在阿尔茨海默病临床治疗中具有巨大应用潜力。去铁胺是一种临床实践中常用的铁螯合剂,可能是治疗阿尔茨海默病的有效方法。有研究对48例阿尔茨海默病患者进行了为期24个月的肌内注射去铁胺单盲试验,与对照组相比,去铁胺将阿尔茨海默病的临床进展率降低了50%,但同时伴随着体质量和食欲下降等不良反应[96]。如前述的维生素E已被发现可以减少谷胱甘肽过氧化物酶4敲除后小鼠大脑中的脂质过氧化,减弱神经元的铁死亡形态,并改善小鼠的认知功能。然而,在随机临床试验中,维生素E对阿尔茨海默病或轻度认知障碍患者的治疗作用并不显著[97]。此外,另一项临床试验发现,与安慰剂相比,维生素E治疗加速了认知能力下降[98]。因此,维生素E在阿尔茨海默病中的使用仍然值得怀疑,需要更多的临床试验来确定其效果。在阿尔茨海默病的治疗中,铁死亡抑制剂Fer-1被证明可以改善体外和体内的神经元死亡和记忆障碍。尽管体内和体外研究已经证明了Fer-1在改善氧化应激和预防铁死亡方面的重要作用,但目前还没有临床试验,其在阿尔茨海默病中的确切疗效及其不良反应需要进一步探索。拉曲匹定是一种抗组胺药,能够通过减少线粒体肿胀和稳定线粒体膜电位作用。在一项纳入中度阿尔茨海默病患者的Ⅱ期临床试验中,拉曲匹定与安慰剂相比显著改善认知功能[99]。然而,一项用拉曲匹定治疗轻度至中度阿尔茨海默病患者中,因拉曲匹定组和安慰剂组之间没有观察到显著差异最终在Ⅲ期临床试验研究时被中止[99]。雷帕霉素是一种雷帕霉素机械靶蛋白的选择性抑制剂,可增加人微管相关蛋白轻链3的水平并抑制雷帕霉素机械靶蛋白信号传导,在体内外能显著改善阿尔茨海默病的病理进程[100]。因此,雷帕霉素机械靶蛋白抑制剂作为靶向治疗阿尔茨海默病的策略能够引起研究者们的广泛关注。姜黄素和白藜芦醇等天然生物活性成分可作为雷帕霉素机械靶蛋白抑制剂来调节线粒体自噬,它们具有结构稳定、安全性高及成本低等优点,具有广泛的临床潜力[101]。然而,大多数关于铁死亡和线粒体自噬调节的研究都是基于细胞和动物模型的临床前研究,这些模型可能无法准确反映对人类的影响。其次关于铜死亡和双硫死亡的机制探索刚刚兴起,临床前研究正处于起步阶段。这些都为靶向铁死亡、线粒体自噬、铜死亡和双硫死亡的药物临床应用带来了挑战。 "

| [1] NGUYEN TT, NGUYEN TTD, NGUYEN TKO, et al. Advances in developing therapeutic strategies for Alzheimer’s disease. Biomed Pharmacother. 2021;139:111623. [2] JORFI M, MAASER-HECKER A, TANZI RE. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023;15(1):6. [3] 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020. doi: 10.1002/alz.12068. [4] BESSEY LJ, WALASZEK A. Management of behavioral and psychological symptoms of dementia. Curr Psychiatry Rep. 2019;21(8):66. [5] TOWER J. Programmed cell death in aging. Ageing Res Rev. 2015;23(Pt A): 90-100. [6] CHAVOSHINEZHAD S, BEIRAMI E, IZADPANAH E, et al. Molecular mechanism and potential therapeutic targets of necroptosis and ferroptosis in Alzheimer’s disease. Biomed Pharmacother. 2023;168:115656. [7] QIU Z, ZHANG H, XIA M, et al. Programmed death of microglia in alzheimer’s disease: autophagy, ferroptosis, and pyroptosis. J Prev Alzheimers Dis. 2023;10(1):95-103. [8] LEMASTERS JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3-5. [9] MATSUDA N, SATO S, SHIBA K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211-221. [10] TSVETKOV P, DETAPPE A, CAI K, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15(7):681-689. [11] DOLMA S, LESSNICK SL, HAHN WC, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285-296. [12] DIXON SJ, LEMBERG KM, LAMPRECHT MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072. [13] DOLL S, PRONETH B, TYURINA YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98. [14] DOLL S, FREITAS FP, SHAH R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-698. [15] TSVETKOV P, COY S, PETROVA B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254-1261. [16] LIU X, NIE L, ZHANG Y, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404-414. [17] LIU X, OLSZEWSKI K, ZHANG Y, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol. 2020;22(4): 476-486. [18] BEDOUI S, HEROLD MJ, STRASSER A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat Rev Mol Cell Biol. 2020;21(11):678-695. [19] NEWTON K, STRASSER A, KAYAGAKI N, et al. Cell death. Cell. 2024;187(2): 235-256. [20] WOODLE ES, KULKARNI S. Programmed cell death. Transplantation. 1998; 66(6):681-691. [21] MIYATA T, NAGY LE. Programmed cell death in alcohol-associated liver disease. Clin Mol Hepatol. 2020;26(4):618-625. [22] WONGPUN J, CHANMANEE T, TOCHARUS J, et al. The potential effects of festidinol treatment against the NLRP3 inflammasome and pyroptosis in D-galactose and aluminum chloride-induced Alzheimer’s-like pathology in mouse brain. Int Immunopharmacol. 2023;119:110181. [23] NONG W, BAO C, CHEN Y, et al. miR-212-3p attenuates neuroinflammation of rats with Alzheimer’s disease via regulating the SP1/BACE1/NLRP3/Caspase-1 signaling pathway. Bosn J Basic Med Sci. 2022;22(4):540-552. [24] ZHANG T, GUAN B, TAN S, et al. Bushen Huoxue Acupuncture Inhibits NLRP1 inflammasome-mediated neuronal pyroptosis in SAMP8 mouse model of Alzheimer’s disease. Neuropsychiatr Dis Treat. 2021;17:339-346. [25] ABDELHADY R, YOUNIS NS, ALI O, et al. Cognitive enhancing effects of pazopanib in D‑galactose/ovariectomized Alzheimer’s rat model: insights into the role of RIPK1/RIPK3/MLKL necroptosis signaling pathway. Inflammopharmacology. 2023;31(5):2719-2729. [26] LI D D, FAN HX, YANG R, et al. Dendrobium nobile lindl. alkaloid suppresses NLRP3-mediated pyroptosis to alleviate LPS-induced neurotoxicity. Front Pharmacol. 2022;13:846541. [27] WANG T, LONG Q, HU Y, et al. miR-181c-5p suppresses neuronal pyroptosis via NLRP1 in Alzheimer’s disease. Behav Brain Res. 2023;447:114387. [28] LI XX, LANG XY, REN TT, et al. Coeloglossum viride var. bracteatum extract attenuates Aβ-induced toxicity by inhibiting RIP1-driven inflammation and necroptosis. J Ethnopharmacol. 2022;282:114606. [29] FANG EF, HOU Y, PALIKARAS K, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401-412. [30] YE X, SUN X, STAROVOYTOV V, et al. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. 2015;24(10):2938-2951. [31] UM JH, SHIN DJ, CHOI SM, et al. Selective induction of Rab9-dependent alternative mitophagy using a synthetic derivative of isoquinoline alleviates mitochondrial dysfunction and cognitive deficits in Alzheimer’s disease models. Theranostics. 2024;14(1):56-74. [32] CHECLER F, GOIRAN T, ALVES DA COSTA C. Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: Implication for neurodegenerative diseases. Autophagy. 2017;13(11): 2004-2005. [33] WANG ZT, LU MH, ZHANG Y, et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell. 2019;18(1):e12860. [34] ZHAO N, ZHANG X, LI B, et al. Treadmill exercise improves PINK1/Parkin-mediated mitophagy activity against Alzheimer’s disease pathologies by upregulated SIRT1-FOXO1/3 axis in APP/PS1 mice. Mol Neurobiol. 2023; 60(1):277-291. [35] BLAGOV AV, GRECHKO AV, NIKIFOROV NG, et al. Role of impaired mitochondrial dynamics processes in the pathogenesis of Alzheimer’s disease. Int J Mol Sci. 2022;23(13):6954. [36] PRADEEPKIRAN JA, RAWAT P, REDDY AP, et al. DDQ anti-aging properties expressed with improved mitophagy in mutant tau HT22 neuronal cells. Mitochondrion. 2024;75:101843. [37] CEN X, CHEN Y, XU X, et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat Commun. 2020;11(1):5731. [38] TANG D, CHEN X, KANG R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107-125. [39] LI J, CAO F, YIN H L, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. [40] PARKER JL, DEME JC, KOLOKOURIS D, et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc. Nat Commun. 2021; 12(1):7147. [41] LI FJ, LONG HZ, ZHOU ZW, et al. System X(c) (-)/GSH/GPX4 axis: an important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13:910292. [42] CHEN C, CHEN W, ZHOU X, et al. Hyperbaric oxygen protects HT22 cells and PC12 cells from damage caused by oxygen-glucose deprivation/reperfusion via the inhibition of Nrf2/System Xc-/GPX4 axis-mediated ferroptosis. PLoS One. 2022;17(11):e0276083. [43] LIU Y, GU W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29(5):895-910. [44] YANG WS, SRIRAMARATNAM R, WELSCH ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317-331. [45] CHEN X, LI J, KANG R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054-2081. [46] HAMBRIGHT WS, FONSECA RS, CHEN L, et al. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8-17. [47] CHEN M, SONG W, CHEN Z, et al. Cerebroprotein hydrolysate attenuates neurodegenerative changes in Alzheimer’s mice model via ferroptosis pathway. Front Pharmacol. 2023;14:1177503. [48] SHAO Y, XU Y, DI H, et al. The inhibition of ORMDL3 prevents Alzheimer’s disease through ferroptosis by PERK/ATF4/HSPA5 pathway. IET Nanobiotechnol. 2023;17(3):182-196. [49] BARUAH P, MOORTHY H, RAMESH M, et al. A natural polyphenol activates and enhances GPX4 to mitigate amyloid-β induced ferroptosis in Alzheimer’s disease. Chem Sci. 2023;14(35):9427-9438. [50] 樊赟,窦润鹏,胡久略,等.茯苓酸通过Nrf2/SLC7A11/GPX4信号通路调控铁死亡改善阿尔茨海默病大鼠认知障碍的研究[J].中国全科医学, 2024,27(2):177-183. [51] STOCKWELL BR, FRIEDMANN ANGELI JP, BAYIR H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273-285. [52] XU Y, LI K, ZHAO Y, et al. Role of Ferroptosis in Stroke. Cell Mol Neurobiol. 2023;43(1):205-222. [53] WANG Y, WU N, LI J, et al. The interplay between autophagy and ferroptosis presents a novel conceptual therapeutic framework for neuroendocrine prostate cancer. Pharmacol Res. 2024;203:107162. [54] ZHOU B, LIU J, KANG R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89-100. [55] MA LL, SUN L, WANG YX, et al. Association between HO‑1 gene promoter polymorphisms and diseases (Review). Mol Med Rep. 2022;25(1):29. [56] LI LB, CHAI R, ZHANG S, et al. Iron exposure and the cellular mechanisms linked to neuron degeneration in adult mice. Cells. 2019;8(2):198. [57] AYTON S, WANG Y, DIOUF I, et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry. 2020;25(11):2932-2941. [58] BELAIDI AA, MASALDAN S, SOUTHON A, et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol Psychiatry. 2022. doi: 10.1038/s41380-022-01568-w. [59] ZHU D, LIANG R, LIU Y, et al. Deferoxamine ameliorated Al(mal)(3)-induced neuronal ferroptosis in adult rats by chelating brain iron to attenuate oxidative damage. Toxicol Mech Methods. 2022;32(7):530-541. [60] BAO WD, PANG P, ZHOU XT, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28(5):1548-1562. [61] YAN C, YANG S, SHAO S, et al. Exploring the anti-ferroptosis mechanism of Kai-Xin-San against Alzheimer’s disease through integrating network pharmacology, bioinformatics, and experimental validation strategy in vivo and in vitro. J Ethnopharmacol. 2024;326:117915. [62] COSTA I, BARBOSA DJ, BENFEITO S, et al. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther. 2023; 244:108373. [63] REICHERT CO, DE FREITAS FA, SAMPAIO-SILVA J, et al. Ferroptosis mechanisms involved in neurodegenerative diseases. Int J Mol Sci. 2020; 21(22):8765. [64] ZHANG H, ZHOU W, LI J, et al. Senegenin rescues pc12 cells with oxidative damage through inhibition of ferroptosis. Mol Neurobiol. 2022;59(11): 6983-6992. [65] WANG M, XUAN T, LI H, et al. Protective effect of FXN overexpression on ferroptosis in L-Glu-induced SH-SY5Y cells. Acta Histochem. 2024;126(1): 152135. [66] 王梦冉.Frataxin介导的铁死亡在阿尔茨海默病细胞模型中的机制研究[D].银川:宁夏医科大学,2023. [67] LONG Q, LI T, ZHU Q, et al. SuanZaoRen decoction alleviates neuronal loss, synaptic damage and ferroptosis of AD via activating DJ-1/Nrf2 signaling pathway. J Ethnopharmacol. 2024;323:117679. [68] LIU ZSJ, TRUONG TTT, BORTOLASCI CC, et al. The potential of baicalin to enhance neuroprotection and mitochondrial function in a human neuronal cell model. Mol Psychiatry. 2024. doi: 10.1038/s41380-024-02525-5. [69] DENG J, ZHUANG H, SHAO S, et al. Mitochondrial-targeted copper delivery for cuproptosis-based synergistic cancer therapy. Adv Healthc Mater. 2024: e2304522. [70] READ AD, BENTLEY RE, ARCHER SL, et al. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021;47:102164. [71] ZHANG P, ZHOU C, REN X, et al. Inhibiting the compensatory elevation of xCT collaborates with disulfiram/copper-induced GSH consumption for cascade ferroptosis and cuproptosis. Redox Biol. 2024;69:103007. [72] LU X, CHEN X, LIN C, et al. Elesclomol loaded copper oxide nanoplatform triggers cuproptosis to enhance antitumor immunotherapy. Adv Sci (Weinh). 2024. doi: 10.1002/advs.202309984. [73] XIONG C, LING H, HAO Q, et al. Cuproptosis: p53-regulated metabolic cell death?. Cell Death Differ. 2023;30(4):876-884. [74] CASTOLDI F, HYVÖNEN MT, DURAND S, et al. Chemical activation of SAT1 corrects diet-induced metabolic syndrome. Cell Death Differ. 2020;27(10): 2904-2920. [75] XUE Q, KANG R, KLIONSKY DJ, et al. Copper metabolism in cell death and autophagy. Autophagy. 2023;19(8):2175-2195. [76] LI X, CHEN X, GAO X. Copper and cuproptosis: new therapeutic approaches for Alzheimer’s disease. Front Aging Neurosci. 2023;15:1300405. [77] CHEN L, MIN J, WANG F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378. [78] PILOZZI A, YU Z, CARRERAS I, et al. A Preliminary study of Cu exposure effects upon Alzheimer’s amyloid pathology. Biomolecules. 2020; 10(3):408. [79] ZHANG Y, ZHOU Q, LU L, et al. Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients. 2023; 15(4):972. [80] LAI Y, LIN C, LIN X, et al. Identification and immunological characterization of cuproptosis-related molecular clusters in Alzheimer’s disease. Front Aging Neurosci. 2022;14:932676. [81] ZHANG E, DAI F, CHEN T, et al. Diagnostic models and predictive drugs associated with cuproptosis hub genes in Alzheimer’s disease. Front Neurol. 2022;13:1064639. [82] 陈桂琳,汤其强.阿尔茨海默病NK细胞与铜死亡的相关基因分析[J/OL].中国组织工程研究,1-9[2024-04-18]. http://kns.cnki.net/kcms/detail/21.1581.R.20240321.0839.006.html. [83] ZHENG P, ZHOU C, DING Y, et al. Disulfidptosis: a new target for metabolic cancer therapy. J Exp Clin Cancer Res. 2023;42(1):103. [84] JIANG H, NECHIPURENKO DY, PANTELEEV MA, et al. Redox regulation of platelet function and thrombosis. J Thromb Haemost. 2024. doi: 10.1016/j.jtha.2024.02.018. [85] CHATZINIKOLAOU PN, MARGARITELIS NV, PASCHALIS V, et al. Erythrocyte metabolism. Acta Physiol (Oxf). 2024;240(3):e14081. [86] PELUCCHI S, STRINGHI R, MARCELLO E. Dendritic spines in alzheimer’s disease: how the actin cytoskeleton contributes to synaptic failure. Int J Mol Sci. 2020;21(3):908. [87] ZHU Y, KONG L, HAN T, et al. Machine learning identification and immune infiltration of disulfidptosis-related Alzheimer’s disease molecular subtypes. Immun Inflamm Dis. 2023;11(10):e1037. [88] FLEMING A, BOURDENX M, FUJIMAKI M, et al. The different autophagy degradation pathways and neurodegeneration. Neuron. 2022;110(6): 935-966. [89] XIE D, SONG C, QIN T, et al. Moschus ameliorates glutamate-induced cellular damage by regulating autophagy and apoptosis pathway. Sci Rep. 2023;13(1):18586. [90] WANG W, GU X, CHENG Z, et al. IKKβ alleviates neuron injury in alzheimer’s disease via regulating autophagy and RIPK1-mediated necroptosis. Mol Neurobiol. 2022;59(4):2407-2423. [91] HUANG Y, XU W, ZHOU R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114-2127. [92] ZHANG D, ZHANG Y, PAN J, et al. Degradation of NLRP3 by p62-dependent-autophagy improves cognitive function in Alzheimer’s disease by maintaining the phagocytic function of microglia. CNS Neurosci Ther. 2023;29(10):2826-2842. [93] KANG R, ZENG L, ZHU S, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24(1):97-108.e104. [94] QUILES DEL REY M, MANCIAS JD. NCOA4-mediated ferritinophagy: a potential link to neurodegeneration. Front Neurosci. 2019;13:238. [95] LI J, LI M, GE Y, et al. β-amyloid protein induces mitophagy-dependent ferroptosis through the CD36/PINK/PARKIN pathway leading to blood-brain barrier destruction in Alzheimer’s disease. Cell Biosci. 2022;12(1):69. [96] CRAPPER MCLACHLAN DR, DALTON AJ, KRUCK TP, et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991; 337(8753):1304-1308. [97] FARINA N, LLEWELLYN D, ISAAC MG, et al. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2017;1(1):Cd002854. [98] LLORET A, BADíA M C, MORA N J, et al. Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis. 2009;17(1):143-149. [99] KE J, TIAN Q, XU Q, et al. Mitochondrial dysfunction: a potential target for Alzheimer’s disease intervention and treatment. Drug Discov Today. 2021;26(8):1991-2002. [100] YANG A JT, FRENDO-CUMBO S, MACPHERSON REK. Resveratrol and metformin recover prefrontal cortex AMPK activation in diet-induced obese mice but reduce BDNF and synaptophysin protein content. J Alzheimers Dis. 2019;71(3):945-956. [101] WANG H, JIANG T, LI W, et al. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol Lett. 2018;282:100-108. |
| [1] | Li Jiagen, Chen Yueping, Huang Keqi, Chen Shangtong, Huang Chuanhong. The construction and validation of a prediction model based on multiple machine learning algorithms and the immunomodulatory analysis of rheumatoid arthritis from the perspective of mitophagy [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(在线): 1-15. |
| [2] | Wang Qiuyue, Jin Pan, Pu Rui . Exercise intervention and the role of pyroptosis in osteoarthritis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1667-1675. |
| [3] | Yin Lu, Jiang Chuanfeng, Chen Junjie, Yi Ming, Wang Zihe, Shi Houyin, Wang Guoyou, Shen Huarui. Effect of Complanatoside A on the apoptosis of articular chondrocytes [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(8): 1541-1547. |
| [4] | Jin Kai, Tang Ting, Li Meile, Xie Yuan. Effects of conditioned medium and exosomes of human umbilical cord mesenchymal stem cells on proliferation, migration, invasion, and apoptosis of hepatocellular carcinoma cells [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1350-1355. |
| [5] | Zhao Nannan, Li Yanjie, Qin Hewei, Zhu Bochao, Ding Huimin, Xu Zhenhua. Changes in ferroptosis in hippocampal neurons of vascular dementia model rats treated with Tongmai Kaiqiao Pill [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1401-1407. |
| [6] | Zhang Mingyang, Yang Xinling. Verbascoside inhibits Erastin-induced ferroptosis of dopaminergic nerve cell line MN9D cells [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1408-1413. |
| [7] | Wan Lingling, Wu Mengying, Zhang Yujiao, Luo Qingqing. Inflammatory factor interferon-gamma affects migration and apoptosis of human vascular smooth muscle cells through pyroptosis pathway [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1422-1428. |
| [8] | Wang Mi, Ma Shujie, Liu Yang, Qi Rui. Identification and validation of characterized gene NFE2L2 for ferroptosis in ischemic stroke [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1466-1474. |
| [9] | Xie Liugang, Cui Shuke, Guo Nannan, Li Aoyu, Zhang Jingrui. Research hotspots and frontiers of stem cells for Alzheimer’s disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(7): 1475-1485. |
| [10] | Liu Zhezhe, Yu Meiqing, Wang Tingting, Zhang Min, Li Baiyan. Troxerutin modulates nuclear factor-kappaB signaling pathway to inhibit brain injury and neuronal apoptosis in cerebral infarction rats [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1137-1143. |
| [11] |
Li Tian, Ren Yuhua, Gao Yanping, Su Qiang.
Mechanism of agomelatine alleviating anxiety- and depression-like behaviors in APP/PS1 transgenic mice #br#
#br#
[J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1176-1182.
|
| [12] | He Guanghui, Yuan Jie, Ke Yanqin, Qiu Xiaoting, Zhang Xiaoling. Hemin regulates mitochondrial pathway of oxidative stress in mouse chondrocytes [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1183-1191. |
| [13] | He Bo, Chen Wen, Ma Suilu, He Zhijun, Song Yuan, Li Jinpeng, Liu Tao, Wei Xiaotao, Wang Weiwei, Xie Jing . Pathogenesis and treatment progress of flap ischemia-reperfusion injury [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1230-1238. |
| [14] | Gao Yang, Qin Hewei, Liu Dandan. ACSL4 mediates ferroptosis and its potential role in atherosclerotic cardiovascular disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1239-1247. |
| [15] | Liu Lingyun, He Guixin, Qin Weibin, Song Hui, Zhang Liwen, Tang Weizhi, Yang Feifei, Zhu Ziyi, Ou Yangbin . Improvement of myocardial injury by traditional Chinese medicine: mitochondrial calcium homeostasis mediates macrophage autophagy and pyroptosis pathway [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(6): 1276-1284. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||