Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (35): 9269-9280.doi: 10.12307/2026.448
Previous Articles Next Articles
Exercise improves neuropathic pain: precision exercise prescription and multimodal synergy advance clinical applications
Guo Feng1, Li Qian2, Hou Chaowen1, Guo Chengji3
- 1Qilu University of Technology, Jining 273100, Shandong Province, China; 2Qufu Traditional Chinese Medicine Hospital, Jining 273100, Shandong Province, China; 3School of Physical Education Sciences, Qufu Normal University, Jining 273165, Shandong Province, China
-
Received:2025-11-18Revised:2026-01-27Online:2026-12-18Published:2026-04-29 -
Contact:Guo Chengji, MS, Professor, School of Physical Education Sciences, Qufu Normal University, Jining 273165, Shandong Province, China -
About author:Guo Feng, MS, Lecturer, Qilu University of Technology, Jining 273100, Shandong Province, China -
Supported by:China Association of Higher Education Project, No. 24TY0211 (to GF)
CLC Number:
Cite this article
Guo Feng, Li Qian, Hou Chaowen, Guo Chengji. Exercise improves neuropathic pain: precision exercise prescription and multimodal synergy advance clinical applications[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(35): 9269-9280.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
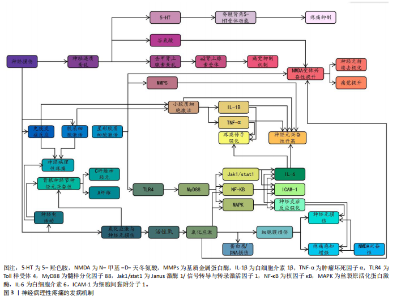
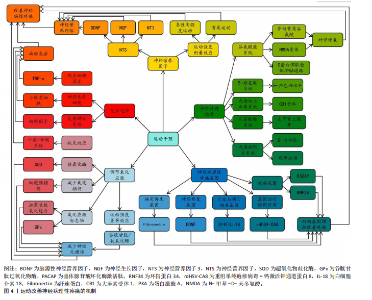
2.1 神经病理性疼痛概述 神经病理性疼痛是由神经损伤或疾病所引发的一种慢性疼痛,主要表现为超出正常生理反应的自发性疼痛和触觉过敏,并难以通过传统药物治疗进行有效控制[3]。相较于其他种类的疼痛,神经病理性疼痛面对外界刺激会产生过度的反应,且伴随连续的自发性疼痛[14],这主要是外周和中枢神经系统损伤、神经炎症、糖尿病神经病变、带状疱疹后遗症、脊髓损伤和癌症治疗后神经损伤等原因所引起[15]。此疼痛为长期痛觉异常,且患者会出现痛觉和触觉过敏。神经病理性疼痛产生的原因较为复杂,神经元兴奋性增加、神经胶质细胞激活、神经递质紊乱、免疫反应和炎症反应等都是发病因素[1]。此外,慢性疼痛还与抑郁、焦虑和睡眠障碍等心理健康问题存在密切关联,这些问题的出现会导致疼痛感知增强,严重降低患者的生活质量[16-17]。目前,神经病理性疼痛的治疗方法包括药物治疗、物理治疗、神经调控技术和心理治疗等,而现有治疗方案会具有明显的不良反应和局限性[3,8]。所以,探索有效的治疗方法理应是疼痛领域研究的重要内容,以期为患者提供更佳的治疗 选择。 2.2 神经病理性疼痛的发病机制 神经病理性疼痛的发病机制涉及神经损伤后多个生理过程的相互作用,包括神经损伤后的神经电活动变化、小胶质细胞和星形胶质细胞的激活、神经递质紊乱、免疫和炎症反应,以及氧化应激和神经元损伤。神经病理性疼痛的发病机制见图3。 2.2.1 神经损伤后的神经电活动变化 神经损伤后的神经电活动变化是产生神经病理性疼痛的一个主要因素。神经损伤发生后,损伤部位神经元和神经网络产生了强烈的电生理变化,导致痛觉信号的异常传递[18]。在外周神经损伤时,受损的神经末梢过度兴奋,使疼痛信号传至中枢神经系统。其中,异常兴奋性通常表现为外周和中枢神经元的持续激活,从而形成一个病理性疼痛回路[4]。外周神经损伤既能导致神经末梢异常兴奋,又能够影响脊髓及脑干等中枢神经系统的功能,进一步促进疼痛信息的传递[19]。此外,感觉神经损伤导致的神经病理性疼痛已经成为全球性的医学难题,主要是因为涉及复杂的神经生理机制和异常疼痛感知[20]。 外周神经损伤后,受损的神经末梢或轴突可能出现超常的自发性放电和异常的疼痛信号传递,这一过程被称为神经元的“异常放电”[21],这一现象常发生于神经损伤后的感觉神经元中,尤其是背根神经节中的A纤维和C纤维神经元,这些神经元的过度敏感性可能引发自发性疼痛以及各种异常的感觉现象[5]。有研究表明,外周神经损伤会使感觉神经元发生超常的自发性放电,进而导致自发性疼痛和过敏反应[22]。同时,损伤部位的神经元对常规刺激过于敏感,表现为触觉过敏或痛觉过敏(例如机械性痛觉过敏和热觉过敏),是神经病理性疼痛患者的典型临床症状表现[6]。 在中枢神经系统中,外周神经的异常放电会经过脊髓背角等神经通路传至大脑,致使中枢神经元亦产生过度激活[23],此类现象与脊髓背角神经元以及上行的痛觉通路损伤紧密相关,损伤后疼痛阈值显著降低,还会形成“痛觉记忆”,虽然原始损伤已得到修复,但疼痛在一段时间内持续存在[23]。中央敏化的机制导致疼痛感知更强烈,致使神经病理性疼痛形成恶性循环[23]。此类神经电活动的异常改变,主要由局部神经损伤引起,同时,亦与中枢神经系统改变有关系,包括神经回路重构和突触传递能力加强[24]。脊髓背角神经元活动提升既提高了对疼痛的反应性,又凭借神经塑性强化了痛觉信号的传递,这成为神经病理性疼痛持续和严重的重要原因[25]。例如在中枢敏化机制中,脊髓背角神经元因钙通道异常促使兴奋性神经递质释放增加,突触传递效率大幅提升;脊髓-"
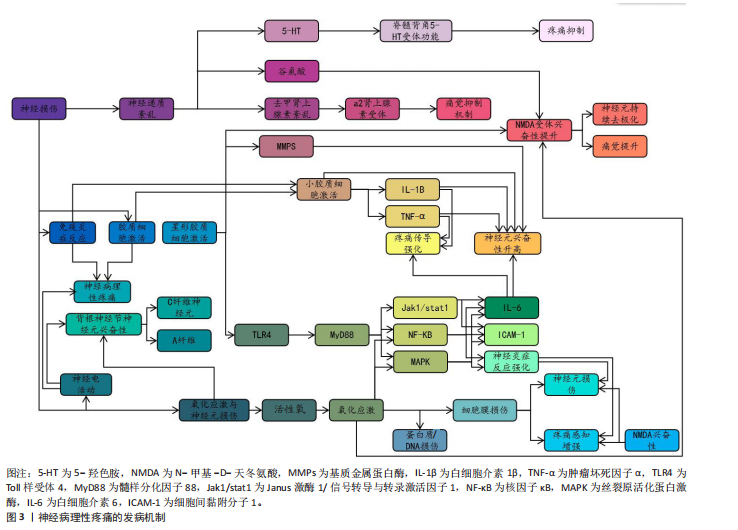
臂旁核通路的短时程可塑性提升也进一步推动了疼痛信号的传递[24]。此外,脊髓小胶质细胞活化通过调控AURKB等分子靶点促进了炎症因子释放和神经可塑性变化,加剧了痛觉超敏反应[26]。研究还证实,卵泡抑素通过激活胰岛素样生长因子1受体信号通路强化了电压门控钠通道1.7亚型功能,直接导致背根神经节神经元兴奋性提升,从而继续维持慢性疼痛状态[27]。 在神经病理性疼痛的外周机制中,背根神经节内不同类型的感觉神经元表现出特征性的异常放电模式:Aβ低阈值机械感受神经元在神经损伤后可出现异位起搏及簇状放电,参与触诱发痛的发生,使感觉信息输入重定向[20,28]; Aδ伤害感受神经元则常表现为短暂高频爆发式放电,并伴有阈下振荡诱发去极化易化;C纤维伤害感受器(包括肽能与非肽能亚群)则以低频持续性自发放电和簇状放电为主,伴随电压门控钠通道1.7亚型、1.8亚型、1.9型、超极化激活环核苷酸门控通道、瞬时受体电位香草酸亚型1等多种离子通道表达上调及电生理激活阈值降低,促使神经瘤或背根神经节局部形成异位起搏点[29-30];此外,轴突断端的脱髓鞘改变及神经元之间的突触旁侧向偶联进一步放大异常电信号,促进机械性痛觉过敏[31]。在中枢层面,持续的外周异常传入活动可在脊髓背角Ⅰ–Ⅱ层诱导“痛觉敏化”现象及N-甲基-D-天冬氨酸受体依赖的长时程增强,该过程涉及突触前谷氨酸与P物质释放增加(与钙通道激活及囊泡循环加速有关),以及突触后NR2B亚基磷酸化、钙调蛋白依赖性蛋白激酶Ⅱ/蛋白激酶C信号通路激活、α-氨基-3-羟基-5-甲基-4-异唑丙酸受体向突触后膜移位等机制。同时,脑源性神经营养因子-酪氨酸激酶受体B信号通路介导的钾氯共转运体2表达下调导致氯离子梯度逆转,从而削弱脊髓抑制性调控,产生“去抑制”效应[32]。小胶质细胞中p38丝裂原活化蛋白激酶与星形胶质细胞中信号转导与转录激活因子3信号通路的激活则共同维持了炎症-代谢耦合基础上的中枢敏化状态[33]。在神经网络层面,延髓头端腹内侧区中“促痛”与“抑痛”神经元活动失衡,蓝斑-脊髓去甲肾上腺素能下行抑制通路功能减弱,以及中脑导水管周围灰质-RVM通路的门控阈值降低,共同导致痛觉敏感性升高[34]。此外,脊髓-臂旁核通路短时程可塑性增强,以及丘脑至前扣带皮质与岛叶皮质的突触传递增益,共同参与痛觉记忆形成及其情绪-认知维度放大[35]。 2.2.2 小胶质细胞和星形胶质细胞的激活 神经损伤不仅直接重塑神经元的电活动模式,还会诱导神经胶质细胞活化,其中以小胶质细胞与星形胶质细胞的响应最为显著[36]。作为中枢神经系统的核心免疫细胞,小胶质细胞在神经损伤后会快速启动应答,深度参与炎症过程的调控,是介导中枢神经系统病理生理变化的关键环节[37],而星形胶质细胞主要承担神经元功能的支持与维持作用,对神经损伤后的响应具有特殊意义,尤其在神经微环境调控与神经功能稳态维持中发挥不可替代的作用[38]。SOFRONIEW等[39]明确指出,中枢神经系统损伤后星形胶质细胞具备强效的响应能力,并强调它在维持神经环境稳定、推动神经修复进程中的核心价值。 神经损伤后的静息态小胶质细胞会快速活化转为效应状态并发生形态重塑,对神经微环境的敏感性提高[40]。同时,活化状态的小胶质细胞会释放大量促炎因子,如肿瘤坏死因子α、白细胞介素1β等,加剧局部乃至全身性的炎症反应[41]。研究证实,小胶质细胞活化是神经病理性疼痛发生的关键因素,不仅参与神经炎症发展过程,还借助疼痛信号传导效率的增强,提升机体的痛觉感知水平[42]。肿瘤坏死因子α、白细胞介素1β等促炎因子被认为是引发并维持神经病理性疼痛的重要媒介,它们通过对脊髓及其他中枢疼痛相关部位的作用,提高神经元兴奋性,最终促使疼痛表型产生[43]。星形胶质细胞在神经损伤后的作用逐步成为关注点。相关研究发现,神经损伤后星形胶质细胞可凭借对神经元支持与修复功能的强化,保证神经系统的基本活动[44]。LIDDELOW等[45]提出星形胶质细胞对神经元存活具有积极促进作用,还可在损伤恢复阶段对神经微环境稳定维持发挥重要作用。但需注意的是,星形胶质细胞过度活化可能诱导白细胞介素1β、基质金属蛋白酶等促炎因子释放,既会加剧神经炎症反应,又可通过调控痛觉传导通路,导致神经病理性疼痛的持续时间延长、强度增强[46]。ZHOU等[47]研究进一步验证,星形胶质细胞通过分泌白细胞介素1β、基质金属蛋白酶等促炎因子参与神经病理性疼痛的形成过程,进而加重神经炎症反应。 小胶质细胞与星形胶质细胞过度活化通过释放炎症递质提高神经元兴奋性,还会通过改变神经元-胶质细胞间的相互作用机制来间接强化痛觉感知[48],这些胶质细胞的异常反应被认为与神经病理性疼痛的慢性化发展及药物抵抗现象密切相关,因此抑制这两类胶质细胞的活化、减少促炎因子释放,可能是缓解神经病理性疼痛的重要解决方案。当前,小胶质细胞在神经系统中的功能研究已取得突破进展。研究发现,静息态小胶质细胞可对脑内微环境变化实施动态监测,在感知刺激信号后会迅速转化为反应状态,并启动免疫应答[49]。其中,p38 丝裂原活化蛋白激酶与核因子κB信号通路会被激活,进一步推动肿瘤坏死因子α、白细胞介素1β、三磷酸腺苷等促炎因子释放,从而对神经元兴奋性造成影响[50]。此外,脑源性神经营养因子可通过特异性受体酪氨酸激酶受体 B上调钾氯共转运体 2的表达水平,调控抑制性神经传递过程,从而介入到疼痛相关的“去抑制”机制[51];同时,趋化因子CX3C配体1/趋化因子CX3C受体1与趋化因子CC配体2/趋化因子CC受体2信号通路在小胶质细胞与初级传入神经元间构成正反馈回路,对急性炎症反应强度及中枢敏化的启动过程进行调控[52]。 在神经系统区域功能方面,脊髓背角具备外周感觉输入的初级放大功能,丘脑与初级体感皮质是感觉信息辨识过程中的关键部分。具体而言,感觉刺激发生时,脊髓背角神经元会开启功能调节,参与外部信号初步增强过程[53];在处理感觉信息任务中,丘脑与初级体感皮质会表现出高度同频的神经活动,这一现象直观体现出二者在感觉信息整合中的协同关系[54]。另外,前扣带皮质与前额叶皮质在情绪-认知放大、疼痛灾难化过程中同样具有关键作用,它们的功能异常可能与情绪障碍及慢性疼痛的发展有着高度关联[55]。小胶质细胞在神经炎症的早期发展过程中就会被活化,通过释放促炎因子参与到神经损伤后的修复过程中;星形胶质细胞则在慢性阶段作用更加显著,主要通过对神经元兴奋性与突触功能进行调控,介导神经可塑性调节[56]。因此,对于胶质细胞的干预策略设计,应考虑疼痛发展阶段和神经区域特点:急性期可针对小胶质细胞的p38 丝裂原活化蛋白激酶/核因子κB、CX3C 趋化因子受体1/趋化因子C-C亚族受体2等信号轴干预,以降低炎症启动阈值;慢性期则需恢复星形胶质细胞中信号转导与转录激活因子3、谷氨酸转运体1等分子的正常功能,同时调控缝隙连接促进神经网络重塑,进而提升镇痛效果的持续性与特异性[57]。 综上所述,神经损伤发生后,小胶质细胞与星形胶质细胞活化并通过释放促炎因子来诱发神经炎症反应,提高神经元兴奋性,最终驱动疼痛信号的传导,成为神经病理性疼痛产生与持续的关键因素。 2.2.3 神经递质紊乱 神经递质紊乱在神经病理性疼痛发展过程中起重要作用。神经递质是神经元之间传递信号的化学物质,它们可协调痛觉的传递和调控[58-59]。在神经损伤后,谷氨酸、5-羟色胺、去甲肾上腺素等神经递质紊乱,形成疼痛恶性循环[58-59]。在神经病理性疼痛中,谷氨酸作为一种兴奋性神经递质起到重要作用。神经损伤后,谷氨酸释放增加促使脊髓背角和大脑神经元持续兴奋,加剧了疼痛信号的感知[60-61]。研究发现,抗癫痫药物通过降低谷氨酸的活性或阻断N-甲基-D-天冬氨酸受体作用来抑制兴奋性传导过度提升是治疗神经病理性疼痛的重要机制之一[62-63]。 5-羟色胺在疼痛调节中同样扮演关键角色。5-羟色胺具备抑制痛觉和在某些条件下加强疼痛感知的双重功能[64]。在神经病理性疼痛中,5-羟色胺的代谢和运输受到损害,而脊髓背角的5-羟色胺受体功能也产生紊乱,导致5-羟色胺对疼痛抑制作用降低,痛觉调控网络平衡被打破,最终产生疼痛[65]。有研究发现,5-羟色胺作用于脊髓背角的5-羟色胺3受体可能会对镇痛药物的疗效产生影响,这说明神经传递的改变可能与神经病理性疼痛的发生紧密相关[66]。此外,去甲肾上腺素等神经递质的异常也与抑郁症、焦虑症等情绪障碍具有高度相关性,可能对疼痛感知具有间接影响关系[67]。研究表明,去甲肾上腺素通过作用于脊髓的α2肾上腺素受体来减轻疼痛,调控痛觉的传导和感知[68]。去甲肾上腺素紊乱通常会导致痛觉抑制机制缺失,加强了痛觉的传导,这也是神经病理性疼痛治疗困难的一个重要因素[68]。 综上所述,神经递质如谷氨酸、5-羟色胺和去甲肾上腺素等在疼痛的传递和调控中扮演重要角色,是神经病理性疼痛发生和持续的关键机制。 2.2.4 免疫和炎症反应 免疫和炎症反应也是神经损伤后产生病理性疼痛的关键机制。神经损伤对神经元功能产生影响的同时,还会激活神经内免疫细胞(如小胶质细胞和星形胶质细胞),促进炎症递质的释放,炎症递质会加强疼痛信号的传递[69]。 小胶质细胞是中枢神经系统的天然免疫细胞,负责清除神经损伤后的废物并对局部炎症反应进行调节[44]。在神经损伤后,小胶质细胞被激活并释放大量促炎因子(如肿瘤坏死因子α、白细胞介素1β、白细胞介素6等),这些因子不仅会使局部产生炎症反应,还会通过与神经元相互作用使神经元兴奋性提升,提高疼痛传导能力[70-71]。研究表明,神经损伤后小胶质细胞激活和促炎因子释放是神经性疼痛发展的重要因素之一[70-71]。 小胶质细胞并非是唯一的激活炎症反应的角色,星形胶质细胞在神经损伤后的免疫反应中也发挥着关键作用。星形胶质细胞在损伤部位活化后促进神经修复与恢复主要依靠分泌细胞因子与基质金属蛋白酶完成;但在神经病理性疼痛情况下,若这些因子释放过度,反而导致神经微环境恶化,进而诱发痛觉感知[72]。已有研究表明,星形胶质细胞内Toll样受体4被激活后,会以髓样分化因子88为媒介,激活核因子κB信号通路,进一步与丝裂原活化蛋白激酶通路、Janus激酶1/信号转导与转录激活因子1通路进行交互,依靠多通路协同调控促炎微环境形成[73]。另有研究表明,α-突触核蛋白及其致病突变体能够在人星形胶质细胞中诱发细胞间黏附分子1与白细胞介素6的表达[74]。此外,神经免疫反应与神经传导通路的重构联系密切。相关研究指出,炎症因子过量释放可强化疼痛相关神经回路的可塑性,导致脊髓背角神经元兴奋性升高,进一步促进疼痛传导并延长持续时间[75],如脊髓背角神经元的突触可塑性变化就是促进疼痛加剧与持续的主要因素[4]。 综上所述,免疫和炎症反应在神经病理性疼痛的发病过程中发挥着不可缺少的作用,神经损伤后的免疫激活通过促进炎症因子释放和提升神经元兴奋性加剧了疼痛感知。 2.2.5 氧化应激和神经元损伤 氧化应激是神经损伤后引发神经病理性疼痛的重要机制。神经损伤发生时,神经对外部刺激产生响应,这一过程会导致活性氧与自由基的生成量迅速上升,发生氧化应激反应[76]。氧化应激通过多种途径对神经元造成损伤,不仅会破坏细胞膜、蛋白质及DNA等结构,还会引起神经细胞膜脂质过氧化,致使细胞膜通透性增加、离子浓度失衡,从而造成细胞内外微环境紊乱[77]。此外,氧化应激还可直接损伤神经细胞的DNA与蛋白质等生物分子,导致细胞功能异常甚至凋亡。相关研究证实,氧化应激不仅在神经损伤的急性阶段发挥作用,还是神经退行性疾病进程中不可忽视的重要影响因素[78-79],损伤机制还会继续诱发神经细胞的病理变化,例如DNA与蛋白质的氧化损伤、细胞凋亡等[79]。 氧化应激水平升高会破坏神经元的正常结构,加剧神经损伤后的功能障碍,自由基与活性氧的过度积累破坏了细胞膜的磷脂结构,致使神经元凋亡以及功能异常,这可能会进一步导致疼痛信号的异常放大[80]。此外,过量的自由基与活性氧会对神经的正常功能造成影响,这也是神经病理性疼痛产生的一个重要原因[81-82]。 在神经病理性疼痛的病理背景下,氧化应激可借助多重机制发挥调控作用,最终加剧痛觉感知水平。文献证实,氧化应激一方面损伤神经细胞结构直接加剧疼痛,另一方面通过提高N-甲基-D-天冬氨酸受体兴奋性,使脊髓与大脑中的神经元更易被激活,最终强化痛觉感知[83]。同时,氧化应激还可激活丝裂原活化蛋白激酶、核因子κB等信号通路,促进神经炎症反应加剧,进而推动神经病理性疼痛发展[84]。相关研究指出,氧化应激可通过生成氧化应激产物、调控蛋白激酶活性、影响胶质细胞功能、激活离子通道等多重途径实现对疼痛反应的调控[85]。 神经病理性疼痛关键发病机制如表1所示。 2.3 运动改善神经病理性疼痛的机制 大量资料证明,运动是一种安全、可控的非药物干预手段,且在减缓神经病理性疼痛方面有着积极作用。从分子层面到系统层面,运动借助多靶点、多通路的协同作用优势实现镇痛效应,其中,精准调控疼痛相关基因的表达是运动发挥改善作用的重要基础。图4描述了运动改善神经病理性疼痛的机制。 2.3.1 运动对神经病理性疼痛相关基因表达的影响 近年文献表明,规律的体育活动能够通过表观遗传调控显著改变基因表达模式[86],这说明运动可能通过重塑基因表达谱参与神经的功能调控。在神经损伤模型中,运动能特异性调控与轴突再生相关的基因网络。例如,HAMED等[87]研究表明糖尿病神经病变大鼠的坐骨神经中纤维连接蛋白基因表达显著降低,而8周有氧运动干预可使纤维连接蛋白基因表达恢复至正常水平,且蛋白表达水平与基因表达呈现同步变化,这说明运动可能通过上调细胞外基质相关基因表达促进神经修复。 有证据显示,神经病理性疼痛大鼠的脊髓背角神经元中Ring finger蛋白34(RNF34)基因异常高表达,而运动干预能抑制Ring finger蛋白34基因表达[13]。从分子机制来看,Ring finger蛋白34通过泛素化作用降解γ-氨基丁酸A型受体的γ2亚基,使得脊髓背角的抑制性神经传递变弱[88],这意味着运动或许能通过双重机制发挥效用:一方面抑制促痛基因Ring finger蛋白34的表达,另一方面通过增加泛素化降解γ-氨基丁酸A型受体的密度,增强疼痛抑制通路。但是,运动调控Ring finger蛋白34的具体信号转导路径目前还不清楚,后续研究需要着重关注运动诱导的微核糖核酸或DNA甲基化修饰机制。 垂体腺苷酸环化酶激活多肽作为一种关键的致痛因子,其基因表达调控与运动镇痛有着密切联系。动物实验发现,向脊髓内注射垂体腺苷酸环化酶激活多肽38会诱发典型的疼痛行为[89],而3周的有氧运动使脊神经结扎大鼠脊髓中垂体腺苷酸环化酶激活多肽mRNA水平显著降低,同时机械性痛觉过敏得到缓解[13]。更有价值的是,MABUCHI等[90]研究表明,垂体腺苷酸环化酶激活多肽基因敲除小鼠在受到N-甲基-D-天冬氨酸刺激时,不会产生机械性痛觉过敏,这说明垂体腺苷酸环化酶激活多肽和N-甲基-D-天冬氨酸受体信号通路存在交互作用。结合运动可降低N-甲基-D-天冬氨酸受体活化的研究证据[91],不难看出,运动可能通过垂体腺苷酸环化酶激活多肽-N-甲基-D-天冬氨酸通路轴发挥多靶点的镇痛效果,但当前研究还未验证运动是如何精确调控该通路中不同节点的基因表达时序,这将是未"
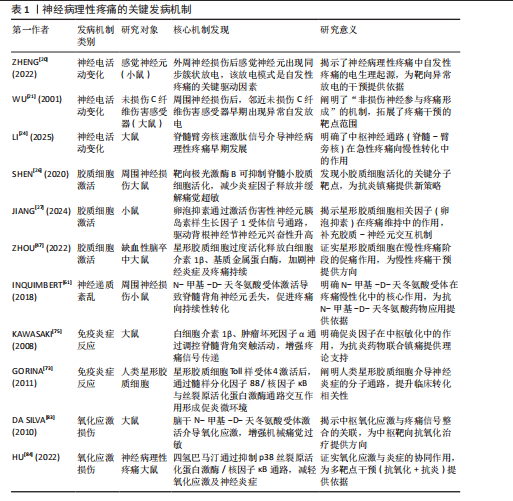
来机制研究的关键方向。 在炎症调控方面,运动展现出了独特的基因调控选择性。在神经损伤小鼠前额叶皮质中,白细胞介素1β和Caspase家族基因1,8,12的过度表达与痛觉过敏密切相关[92],而运动训练能显著抑制促炎和促凋亡基因的表达[93],这表明运动可能通过调控神经免疫的基因表达网络来实现抗炎镇痛。运动对神经营养因子的正向调控作用也很明显,例如能增加脑源性神经营养因子和神经营养因子3的mRNA表达,促进外周神经损伤后的轴突再生[94],这些结果共同构成了运动调控基因表达的双向模型:既抑制疼痛相关基因,又激活神经修复基因。另外,基因治疗与运动干预相结合展现出了重要的应用前景。最新文献显示,采用rdHSV-CA8基因治疗能够通过激活Kv7钾通道降低神经元兴奋性,从而显著减轻神经病理性疼痛[95],这说明将运动诱导的内源性基因调控与外源性基因治疗相结合,可能会产生协同治疗效果。 综上所述,运动通过双向基因调控来减轻神经病理性疼痛:一方面抑制促痛基因的表达,减少泛素化对γ-氨基丁酸A型的降解以及N-甲基-D-天冬氨酸的敏化;另一方面激活神经再生基因以促进修复,同时调控白细胞介素1β/Caspase轴,重塑神经免疫稳态。基因治疗联合运动展现出了协同镇痛的潜力。当前存在的瓶颈主要有:①机制研究大多局限于动物模型,人类的表观遗传特征以及运动参数的推进有待确定;②基因编辑与运动干预的协同方案需要验证。未来需要结合单细胞测序来解析运动调控的细胞特异性,构建“基因靶点-精准运动”的转化路径。 2.3.2 运动对神经病理性疼痛相关神经递质释放的影响 在探索运动改善神经病理性疼痛的过程中,内源性阿片系统长期以来被视为一条关键的镇痛路径[96]。神经化学方面的证据表明,运动能够促使内源性阿片类物质释放,产生明确的止痛作用,这一点已得到大量实验数据的支持。研究进一步证实,运动可诱发阿片系统的迅速响应,响应程度与运动强度紧密关联,且无性别差异[97]。然而,此类短期激活如何逐步转化为阿片系统长期的功能重构,其内在机制仍不清晰。STAGG课题组在神经病理性疼痛动物模型中观察到,定期运动不仅能即时促进内源性阿片释放,还可使其活性在较长时间内处于较高状态[9],这提示运动可能借助神经可塑性,实现对阿片系统的持续动员。临床层面的证据也加强了这一观点:HUGHES等[98]报道罹患癌相关神经病理性疼痛的患者在接受抗阻训练后,由β-内啡肽介导的运动诱导镇痛效应与中枢阿片受体激活以及下行抑制通路功能改善有明确关联。目前研究仍存在明显短板,例如不同运动类型(如耐力性与抗阻性训练)是否对阿片受体各亚型产生差异化调控,尚缺乏系统对比;未来有必要采用实时微透析等动态监测手段,深入揭示运动强度与阿片释放之"
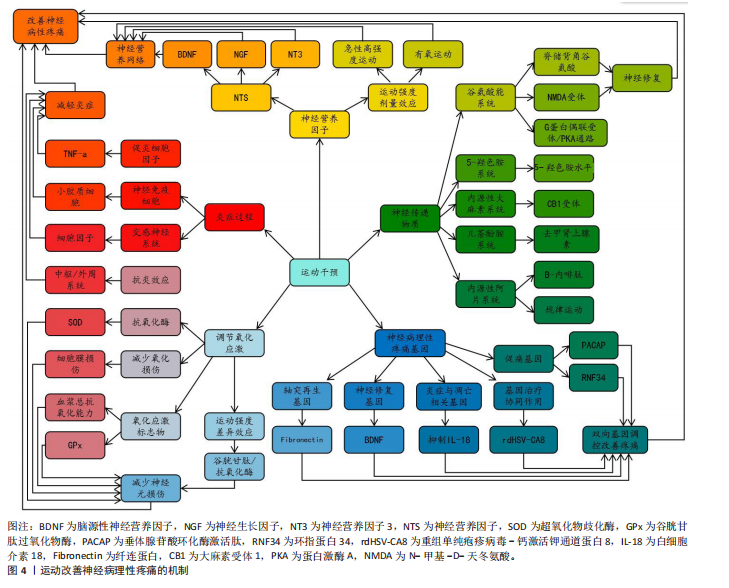
间的实时关系。 尽管神经递质释放变化并非神经病理性疼痛的根本病因,其在疼痛信号调节中却发挥着不可忽视的调控功能[99]。已有研究指出,钙通道α2δ亚基表达上调引发谷氨酸释放过量,是加剧疼痛信号传递的重要环节[100],而运动则可能借助双路径策略缓解这一异常。在动物实验中,神经病理性疼痛大鼠的脊髓背角区域谷氨酸释放明显增多[101],而经过游泳运动干预后脑脊液内谷氨酸浓度显著下降[102],这表明运动或许通过调节突触前钙通道功能,减少谷氨酸的病理释放。另一方面,SLUKA等[91]研究提出,强制性跑步运动能够通过阻碍N-甲基-D-天冬氨酸受体NR1亚基的磷酸化进程,降低突触后信号传递效能;MARTINS等[14]进一步阐明,游泳运动可通过G蛋白偶联受体-蛋白激酶A信号途径,对抗谷氨酸所引发的痛觉过敏。这些发现共同说明,运动对谷氨酸能系统的影响同时覆盖突触前与突触后环节。但需要指出的是,其中涉及的具体分子路径仍有争议,后续研究可借助条件性基因敲除模型,进一步厘清不同通路在镇痛中的实际贡献。 儿茶酚胺系统在运动介导的镇痛过程中同样具有重要地位。动物研究显示,运动激活交感神经系统后,会促使去甲肾上腺素释放,该递质随后作用于脊髓α2肾上腺素能受体,引发下行性抑制反应[103]。值得关注的是,这一效应可能与内源性大麻素系统存在功能交汇:GALDINO等[104]报道大麻素受体参与对运动镇痛的调控;MARTINS等[14]则发现CB1受体拮抗剂AM281能够特异性阻断雄性小鼠由运动引发的镇痛反应,此类性别差异提示不同神经调质系统之间的协同方式可能因人而异,也为临床实施个体化干预策略提供了思路。在单胺类递质中,5-羟色胺的调节功能尤为突出。BOBINSKI等[105]研究表明,神经损伤会导致脑干区5-羟色胺水平下降,同时其转运体表达上升,而为期2周的跑步机训练则能逆转上述异常;若使用药物抑制5-羟色胺合成,运动的镇痛作用会显著减弱。LIMA等[106]进一步补充,运动能够通过抑制背腹侧髓质区血清素转运体的过度表达,维持突触间隙中5-羟色胺的有效浓度。总体来看,现有证据提示,运动可能通过阿片类、儿茶酚胺、大麻素以及5-羟色胺等多个系统的协同参与,重新塑造疼痛调控网络。无论是耐力型还是力量型运动,均有助于提高这些递质系统的整体活性,共同推动痛阈上升[98]。 综上,运动通过多靶点调控神经递质释放减缓神经病理性疼痛,然而,尚需解决以下问题:①不同运动参数对特定递质的调控特异性;②递质交互作用的时空动态特征;③临床转化中运动处方的个性化设计策略。未来研究应梳理电生理与多组学技术,动态解析运动干预下神经递质网络的适应性改变。 2.3.3 运动对神经病理性疼痛相关神经营养因子的影响 神经营养因子是调控神经元存活与功能的关键蛋白家族。大量体内(动物模型)和体外研究表明,神经营养因子通过调控神经元的生长、维持及凋亡过程,在神经的发育与修复中发挥关键作用[107]。神经营养因子家族包括神经生长因子、脑源性神经营养因子、神经营养因子3和神经营养因子4等成员,这些因子虽具备相似的三维结构,但在功能表达上存在时空特异性差异[108],这说明神经的可塑性调控可能依赖于不同神经营养因子的协同作用。 在神经损伤修复研究领域,神经生长因子是目前研究最为深入的神经营养因子之一。动物实验证实,在坐骨神经损伤模型中,神经生长因子不仅能增强施万细胞清除髓鞘碎片的能力,还能通过信号转导与转录激活因子3磷酸化通路显著促进神经再生[109]。临床研究也取得了支持性证据:急性脑出血患者经神经生长因子治疗后,Barthel指数改善率达到31.7%,且未发现严重不良反应[110]。然而,神经生长因子在不同神经损伤阶段的最佳剂量-效应关系仍不明确,长期使用的安全性也有待验证,这些因素制约了临床转化进程。近年来,研究视野逐步扩展到运动干预对神经营养因子网络的调控作用。研究表明,规律运动能显著促进神经营养因子的表达,中枢神经系统内的脑源性神经营养因子浓度可比基线水平提高1.8倍[111]。特别值得注意的是,运动引起的神经营养因子变化表现出明显的性别差异。在糖尿病前期小鼠中,8周自主跑轮运动使坐骨神经TrkB磷酸化水平提升55%,机械痛阈也从3.2 g提高至6.5 g[112]。这些发现提示,运动可能通过重塑神经营养因子网络来缓解神经病理性疼痛,但具体分子机制仍需深入探讨。 在剂量效应关系方面,人体研究提供了重要数据:高强度运动(80% VO2 max)可使血清脑源性神经营养因子浓度短时间内上升72%,而中等强度运动(50% VO2 max)仅引起35%的增长[113],这种强度依赖性效应在疼痛调控中具有重要意义。ALMEIDA等[10]发现,4周有氧运动使神经病理性疼痛小鼠背根神经节脑源性神经营养因子表达降低38%,机械痛敏阈值由2.3 g升至5.1 g。不过,关于神经营养因子在疼痛调控中的作用方向仍存在分歧,有研究认为运动通过降低神经生长因子/脑源性神经营养因子表达来抑制伤害性神经元异常发芽[11]。这些矛盾结果表明,神经营养因子可能具有“双刃剑”特性,最终效应取决于局部微环境的动态平衡。 基于现有证据,未来研究应着重于以下方向:建立运动参数与神经营养因子表达特征的动态关联模型;阐明神经营养因子在神经修复与疼痛调控中的浓度阈值效应;开发针对神经营养因子信号通路的运动-药物协同治疗方案。这些研究的推进将为神经病理性疼痛的精准干预提供理论依据。 2.3.4 运动对神经病理性疼痛相关炎症过程的影响 神经病理性疼痛作为神经损伤引发的慢性疼痛类型,炎症反应激活是核心机制之一。神经损伤会推动炎症递质的表达与生成,尤以小鼠模型中肿瘤坏死因子α、白细胞介素 1β等细胞因子升高最为显著[12],这些细胞因子的累积与小胶质细胞活化紧密关联,进而形成 “炎症持续激活”的恶性循环,最终诱发神经病理性疼痛。近年研究证实,运动干预可在一定程度上调控这一病理过程,减轻神经病理性疼痛症状。 多个研究结果表明,运动对小鼠的炎症与免疫反应具有调节作用,能够帮助中枢及外周神经中细胞因子、神经免疫递质水平恢复到损伤前状态 [114],该调控作用不仅作用于急性炎症阶段,长期运动还能在一定程度上改善神经免疫功能。具体来讲,运动可通过降低促炎细胞因子水平、上调抗炎细胞因子表达缓解神经炎症反应[115]。其中,有氧运动已被证实可明显减轻糖尿病等慢性疾病伴随的炎症反应[116]。更重要的是,运动对神经病理性疼痛的缓解作用可能与交感神经激活相关联:运动减少了小胶质细胞活化及细胞因子释放,减轻了神经病理性疼痛症状[117],这说明运动可能通过调控免疫反应起到抗炎作用,但目前仍缺少有关 “不同运动类型如何影响不同阶段神经损伤” 的具体内容,因此后续研究应围绕运动干预的长期效果进行评估,明晰不同运动形式的各方面效果差异。 神经病理性疼痛大鼠经过 3 周跑步机运动后,脑脊液中肿瘤坏死因子α水平显著降低 [12],这说明运动不仅能调节神经免疫微环境,还可通过减少细胞因子表达缓解痛觉感受。CHEN等[118]也发现神经病理性疼痛大鼠经21 d游泳或跑步机运动后,坐骨神经中肿瘤坏死因子α与白细胞介素1β水平显著下降。 这些研究结果证实,运动通过降低炎症相关细胞因子表达,在一定程度上抑制了神经病理性疼痛的发生发展,可见运动作为一种有效抗炎手段,既能缓解神经病理性疼痛,还可能通过多机制改善神经免疫功能。但目前仍需进一步探究运动模式、强度、时长等因素对神经病理性疼痛不同机制的影响,未来研究应聚焦运动在临床神经病理性疼痛管理中的实际应用,尤其需明确不同类型运动对疼痛缓解的长期效果。 2.3.5 运动对神经病理性疼痛相关氧化应激的影响 氧化应激是神经性疼痛发展的机制之一[119]。神经含有大量磷脂,容易受到氧化应激的影响[120]。氧化应激与神经病理性疼痛的关系日益得到关注,许多文献表明运动在减缓氧化应激、促进疼痛感受方面具备积极作用。 运动能够增强抗氧化酶活性、减轻氧化损伤,并提高组织对抗自由基的能力,从而发挥有益调节功能。此类研究目前多集中于雄性动物模型。以PINHO团队[121]的工作为例,研究证实运动可以通过提高超氧化物歧化酶、谷胱甘肽过氧化物酶及过氧化氢酶等关键抗氧化酶的活性,有效缓解氧化应激带来的损伤。GHANBARI[122]研究同样表明,规律有氧运动能够抑制脂质过氧化反应,从而降低氧化损伤程度。然而需要认识到,运动对不同氧化应激标志物的影响并不完全一致。尽管运动普遍提升抗氧化酶活性,但调控效果在不同物种间存在差异,例如DA SILVA等[123]发现,运动促进小鼠肝脏中某些氧化应激标志物的表达,不同运动模式对各类标志物的影响程度各不相同。FULLE等[124]进一步提出,活性氧本身可通过激活特定信号通路,参与调节肌肉组织中抗氧化酶的活性。 运动对氧化应激的调节效果还可能受到机体基础氧化状态与运动强度的共同调节。研究发现,中等强度跑步机训练虽未显著改变大鼠基础谷胱甘肽水平,但在经过特定药物诱导后,运动能有效恢复谷胱甘肽水平[125],这说明在特定条件下,运动对氧化应激相关酶的恢复作用较为显著,但在不同实验环境中,运动与氧化应激的关系可能呈现非线性特征。SAFAKHAH等[12]报道,神经病理性疼痛模型大鼠经过3周跑步机训练后,血浆总抗氧化能力显著提升。ROSTAMI等[126]研究也表明,游泳运动能够逆转三叉神经损伤引起的谷胱甘肽过氧化物酶活性下降。 尽管上述研究结果形成了较为全面的论述,但急性运动对氧化应激的影响仍有不同观点。急性运动可能诱导活性氧生成瞬时提升氧化应激水平,以及增加肌肉痛觉感受器的敏感度[127];而长期规律运动则被证实可降低氧化应激相关疾病发生率,并借助激活抗炎反应减缓慢性疼痛[127]。在短期运动对疼痛的影响方面,未形成统一研究结论:部分研究认为短期运动可能使疼痛加重[128],而有研究则认为短期运动可短暂降低痛觉敏感性[9]。这种结果差异可能源于对慢性疼痛的不同病理机制的探讨。未来应进一步深入探究运动强度、持续时间及频率对神经病理性疼痛和氧化应激的多重影响机制。 现有研究为神经病理性疼痛的非药物干预提供了新思路,后续工作应重点关注运动强度、运动形式与不同疼痛模型之间的特异性对应关系。运动干预神经病理性疼痛研究如表2所示。 2.4 不同运动方式与强度对炎症与氧化应激通路的差异作用 神经病理性疼痛的发生发展涉及炎症反应与氧化应激之间的恶性循环。其中,小胶质细胞和星形胶质细胞活化、核因子κB信号通路及炎症小体激活、促炎因子释放等炎症过程,与活性氧蓄积、线粒体功能异常及抗氧化防御系统失调等氧化应激改变相互促进,共同加剧疼痛感知和神经功能损伤。在这一过程中,运动干预通过骨骼肌分泌因子-全身代谢通路-神经免疫网络的级联反应,发挥运动方式与强度依赖特征的双向调节作用。 中低强度有氧运动能够通过多靶点机制同步改善炎症和氧化应激状态。实验显示中低强度有氧运动可上调过氧化物酶体增殖物激活受体γ共激活因子1α、腺苷单磷酸活化蛋白激酶、沉默信息调节因子1及脑源性神经营养因子等重要调控分子,促进线粒体生物合成与能量代谢,同时抑制NOD样受体热蛋白结构域相关蛋白3炎症小体表达,激活核因子E2相关因子2与cAMP反应元件结合蛋白通路,并遏制核因子κB的过度活化,从而增强抗氧化防御、减轻神经炎症并改善突触可塑性,最终缓解抑郁等共病症状[129]。在帕金森病模型中,运动通过调节Toll样受体/髓样分化因子88/核因子κB抑制蛋白α信号轴,降低白细胞介素1β与肿瘤坏死因子α水平,提高白细胞介素10与转化生长因子β等抗炎因子水平,同时抑制凋亡相关斑点样蛋白与NOD样受体热"
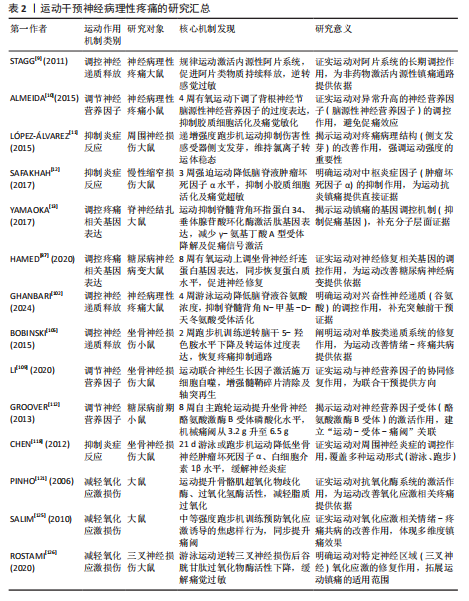

蛋白结构域相关蛋白3炎症小体复合物形成,减少神经元凋亡并促进神经修复[130]。此外,有氧运动还可通过激活骨骼肌中腺苷单磷酸活化蛋白激酶/过氧化物酶体增殖物激活受体γ共激活因子1α信号轴来恢复脂肪酸氧化与葡萄糖代谢平衡,促进肌肉因子释放,进而维持脑内能量稳态,减缓全身性炎症反应[131]。在抗氧化方面,运动诱导过氧化物酶体增殖物激活受体γ共激活因子1α、腺苷单磷酸活化蛋白激酶、沉默信息调节因子1激活可显著增强超氧化物歧化酶、过氧化氢酶与谷胱甘肽过氧化物酶等关键抗氧化酶的表达,改善线粒体功能与氧化损伤[132]。高强度间歇训练则在调节氧化应激方面发挥出独特价值表现。研究发现,高强度间歇训练可显著激活核因子E2相关因子2通路,强化抗氧化酶活性并降低肝脏内质网应激反应,该作用在代谢异常模型中尤为突 出[133]。值得一提的是,高强度间歇训练可能在炎症高度活跃状态下加重急性痛敏反应,需谨慎考虑并应用。身心整合类运动(如瑜伽)通过结合身体活动、呼吸调节与认知训练,对炎症状态和自主神经平衡发挥积极作用。多项研究结果证实,瑜伽干预可明显改善乳腺癌患者的生活质量,缓解疲劳、睡眠障碍以及抑郁和焦虑症状,作用机制可能与调节心率变异性、增强迷走神经张力及降低神经内分泌应激水平有关[134]。长期瑜伽练习者在静息状态下具有更高的迷走神经张力,揭示了此类运动对自主神经平衡的恢复有帮助[135]。 针对不同临床表现,运动处方需个体化调整:对于促炎细胞因子升高伴急性痛敏的患者,建议首选低至中等强度有氧运动(每周时长递增约10%),慎用高强度间歇训练,同时配合身心整合干预以降低神经内分泌应激,待症状稳定后,可逐步加入中等强度抗阻训练。研究证实,低强度有氧运动可通过增强脑干5-羟色胺神经传递,抑制白细胞介素1β与肿瘤坏死因子α等促炎因子,有效缓解神经病理性疼痛[105]。 身心整合方法(如认知行为疗法)可作为重要辅助手段,改善疼痛相关的功能障碍[136]。中等强度抗阻训练通过促进抗炎因子白细胞介素6与内源性镇痛物质(如β-内啡肽)释放,调节疼痛传导通路[137]。 对于老年患者,建议以抗阻训练为核心,结合有氧训练(每周至少2次抗阻训练+150 min有氧运动),并进行身心整合干预。研究表明,抗阻与有氧结合训练可显著降低C-反应蛋白、白细胞介素6与肿瘤坏死因子α等炎症标志物,在健康老年人群中抗炎效果尤为明显[138]。对于表现为痛觉过敏与情绪障碍的患者,初期应以身心整合训练与中低强度有氧运动为主,待痛阈提升后再逐步引入抗阻训练,避免训练强度大幅波动。实验显示,深慢呼吸等放松技巧可有效提高痛阈,降低交感神经兴奋性,改善情绪调节能力[139]。 综上所述,不同运动方式与强度对炎症与氧化应激通路的调节存在明确的“剂量-方式-表型”匹配关系:低中强度有氧运动与身心整合干预更有利于急性炎症抑制与整体氧化还原稳态重建;而抗阻训练与经过筛选的高强度间歇训练在慢性期功能恢复与抗氧化适应中贡献更为突出。以表型分层为基础、结合动态监测的处方化策略,有助于实现个体化、机制导向的镇痛与功能改善。"

| [1] DIBONAVENTURA MD, SADOSKY A, CONCIALDI K, et al. The prevalence of probable neuropathic pain in the US: results from a multimodal general-population health survey. J Pain Res. 2017;10:2525-2538. [2] 吴大胜,陶蔚,朱谦.神经病理性疼痛评估与管理中国指南(2024版)[J].中国疼痛医学杂志, 2024,30(1):5-14. [3] 邵仪,蓝心,陈运滏,等.神经病理性疼痛的研究进展[J].疑难病杂志,2024,23(4):491-495. [4] WOOLF CJ, SALTER MW. Neuronal plasticity: increasing the gain in pain. Science. 2000; 288(5472):1765-1769. [5] COSTIGAN M, SCHOLZ J, WOOLF CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009; 32:1-32. [6] JENSEN TS, FINNERUP NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 2014;13(9):924-935. [7] INOUE K, TSUDA M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci. 2018; 19(3):138-152. [8] 郭楠楠,郭雪娇,冯智英.神经病理性疼痛分层治疗进展[J].中国疼痛医学杂志,2022, 28(1):49-52. [9] STAGG NJ, MATA HP, IBRAHIM MM, et al. Regular exercise reverses sensory hypersensitivity in a rat neuropathic pain model: role of endogenous opioids. Anesthesiology. 2011;114(4):940-948. [10] ALMEIDA C, DEMAMAN A, KUSUDA R, et al. Exercise therapy normalizes BDNF upregulation and glial hyperactivity in a mouse model of neuropathic pain. Pain. 2015;156(3):504-513. [11] LÓPEZ-ÁLVAREZ VM, MODOL L, NAVARRO X, et al. Early increasing-intensity treadmill exercise reduces neuropathic pain by preventing nociceptor collateral sprouting and disruption of chloride cotransporters homeostasis after peripheral nerve injury. Pain. 2015;156(9):1812-1825. [12] SAFAKHAH HA, MORADI KOR N, BAZARGANI A, et al. Forced exercise attenuates neuropathic pain in chronic constriction injury of male rat: an investigation of oxidative stress and inflammation. J Pain Res. 2017;10:1457-1466. [13] YAMAOKA S, OSHIMA Y, HORIUCHI H, et al. Altered Gene Expression of RNF34 and PACAP Possibly Involved in Mechanism of Exercise-Induced Analgesia for Neuropathic Pain in Rats. Int J Mol Sci. 2017;18(9):1962. [14] MARTINS DF, SITENESKI A, LUDTKE DD, et al. High-Intensity Swimming Exercise Decreases Glutamate-Induced Nociception by Activation of G-Protein-Coupled Receptors Inhibiting Phosphorylated Protein Kinase A. Mol Neurobiol. 2017;54(7):5620-5631. [15] 张泾纬,刘尧,高灿.神经病理性疼痛与认知功能障碍共病的细胞和分子机制研究进展[J].中国疼痛医学杂志,2025,31(3):196-201. [16] 王笑珂,舒晴.慢性疼痛与负性情绪的神经交互机制及综合干预策略研究进展[J].中山大学学报(医学科学版),2025,46(2):197-209. [17] VINALL J, PAVLOVA M, ASMUNDSON GJ, et al. Mental Health Comorbidities in Pediatric Chronic Pain: A Narrative Review of Epidemiology, Models, Neurobiological Mechanisms and Treatment. Children (Basel). 2016;3(4):40. [18] 吴志伟,宋朋飞,朱清广,等.神经病理性疼痛机制研究进展[J].河北医科大学学报,2018, 39(9):1095-1100. [19] RICHNER M, ULRICHSEN M, ELMEGAARD SL, et al. Peripheral nerve injury modulates neurotrophin signaling in the peripheral and central nervous system. Mol Neurobiol. 2014;50(3):945-970. [20] ZHENG Q, XIE W, LÜCKEMEYER DD, et al. Synchronized cluster firing, a distinct form of sensory neuron activation, drives spontaneous pain. Neuron. 2022;110(2):209-220.e6. [21] WU G, RINGKAMP M, HARTKE TV, et al. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci. 2001;21(8):RC140. [22] SONG X, WANG Y, YANG W, et al. Abnormal Spontaneous Discharges of Primary Sensory Neurons and Pain Behavior in a Rat Model of Vascular Dementia. Int J Mol Sci. 2023;24(12): 10198. [23] WOOLF CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2-S15. [24] LI Y, HA NT, LI J, et al. Tachykinin signaling in the right parabrachial nucleus mediates early-phase neuropathic pain development. Neuron. 2025;113(4):605-619.e6. [25] TANSLEY S, GU N, GUZMÁN AU, et al. Microglia-mediated degradation of perineuronal nets promotes pain. Science. 2022;377(6601):80-86. [26] SHEN Y, DING Z, MA S, et al. Targeting aurora kinase B alleviates spinal microgliosis and neuropathic pain in a rat model of peripheral nerve injury. J Neurochem. 2020;152(1):72-91. [27] JIANG BC, LING YJ, XU ML, et al. Follistatin drives neuropathic pain in mice through IGF1R signaling in nociceptive neurons. Sci Transl Med. 2024;16(769):eadi1564. [28] DEVOR M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res. 2009;196(1):115-128. [29] AMIR R, MICHAELIS M, DEVOR M. Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci. 2002; 22(3):1187-1198. [30] VON HEHN CA, BARON R, WOOLF CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012; 73(4):638-652. [31] FREIHA J, RIACHI N, CHALAH MA, et al. Paroxysmal Symptoms in Multiple Sclerosis-A Review of the Literature. J Clin Med. 2020;9(10):3100. [32] FERRINI F, PEREZ-SANCHEZ J, FERLAND S, et al. Differential chloride homeostasis in the spinal dorsal horn locally shapes synaptic metaplasticity and modality-specific sensitization. Nat Commun. 2020;11(1):3935. [33] JI RR, SUTER MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. [34] CHEN Q, HEINRICHER MM. Shifting the Balance: How Top-Down and Bottom-Up Input Modulate Pain via the Rostral Ventromedial Medulla. Front Pain Res (Lausanne). 2022;3:932476. [35] DENG J, ZHOU H, LIN JK, et al. The Parabrachial Nucleus Directly Channels Spinal Nociceptive Signals to the Intralaminar Thalamic Nuclei, but Not the Amygdala. Neuron. 2020;107(5):909-923.e6. [36] 刘健,李伟彦.神经胶质细胞与神经病理性疼痛[J].医学研究生学报,2009,22(2):209-212. [37] 闫立成.小胶质细胞激活在镧致神经元损伤中的作用及机制研究[D].沈阳:中国医科大学, 2019. [38] 邢文晓,罗富成,吕涛.中枢神经系统中的星形胶质细胞通过多种机制调控髓鞘发育和再生[J].生物化学与生物物理进展,2025, 52(7):1792-1803. [39] SOFRONIEW MV, VINTERS HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010; 119(1):7-35. [40] VIDAL-ITRIAGO A, RADFORD RAW, ARAMIDEH JA, et al. Microglia morphophysiological diversity and its implications for the CNS. Front Immunol. 2022;13:997786. [41] DISABATO DJ, QUAN N, GODBOUT JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139 Suppl 2(Suppl 2):136-153. [42] ELLIS A, BENNETT DL. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111(1):26-37. [43] NADEAU S, FILALI M, ZHANG J, et al. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1β and TNF: implications for neuropathic pain. J Neurosci. 2011;31(35):12533-12542. [44] 付海鑫,王慧影,张良,等.神经变性疾病与神经炎症:小胶质细胞和星形胶质细胞的作用[J].中风与神经疾病杂志,2023,40(9):859-864. [45] LIDDELOW SA, GUTTENPLAN KA, CLARKE LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481-487. [46] ZHAO H, ALAM A, CHEN Q, et al. The role of microglia in the pathobiology of neuropathic pain development: what do we know?. Br J Anaesth. 2017;118(4):504-516. [47] ZHOU M, ZHANG T, ZHANG X, et al. Effect of Tetrahedral Framework Nucleic Acids on Neurological Recovery via Ameliorating Apoptosis and Regulating the Activation and Polarization of Astrocytes in Ischemic Stroke. ACS Appl Mater Interfaces. 2022;14(33): 37478-37492. [48] WIESELER-FRANK J, MAIER SF, WATKINS LR. Glial activation and pathological pain. Neurochem Int. 2004;45(2-3):389-395. [49] NIMMERJAHN A, KIRCHHOFF F, HELMCHEN F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314-1318. [50] PARAJULI B, SONOBE Y, KAWANOKUCHI J, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation. 2012;9:268. [51] PORCHER C, MEDINA I, GAIARSA JL. Mechanism of BDNF Modulation in GABAergic Synaptic Transmission in Healthy and Disease Brains. Front Cell Neurosci. 2018;12:273. [52] CLARK AK, MALCANGIO M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front Cell Neurosci. 2014;8:121. [53] PANTOJA J, RIBEIRO S, WIEST M, et al. Neuronal activity in the primary somatosensory thalamocortical loop is modulated by reward contingency during tactile discrimination. J Neurosci. 2007;27(39):10608-10620. [54] PAIS-VIEIRA M, LEBEDEV MA, WIEST MC, et al. Simultaneous top-down modulation of the primary somatosensory cortex and thalamic nuclei during active tactile discrimination. J Neurosci. 2013;33(9):4076-4093. [55] VAN NOORT J, VERBRUGGE S, GOOSEN N, et al. Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proc Natl Acad Sci U S A. 2004;101(18):6969-6974. [56] HORSCHITZ S, LAU T, SCHLOSS P. Glycine residues G338 and G342 are important determinants for serotonin transporter dimerisation and cell surface expression. Neurochem Int. 2008;52(4-5): 770-775. [57] BRAMINI M, CHIACCHIARETTA M, ARMIROTTI A, et al. An Increase in Membrane Cholesterol by Graphene Oxide Disrupts Calcium Homeostasis in Primary Astrocytes. Small. 2019;15(15):e1900147. [58] KAUR J, GHOSH S, SAHANI AK, et al. Mental imagery training for treatment of central neuropathic pain: a narrative review. Acta Neurol Belg. 2019;119(2):175-186. [59] PASERO C. Pathophysiology of neuropathic pain. Pain Manag Nurs. 2004;5(4 Suppl 1):3-8. [60] WENG HR, CHEN JH, CATA JP. Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience. 2006; 138(4):1351-1360. [61] INQUIMBERT P, MOLL M, LATREMOLIERE A, et al. NMDA Receptor Activation Underlies the Loss of Spinal Dorsal Horn Neurons and the Transition to Persistent Pain after Peripheral Nerve Injury. Cell Rep. 2018;23(9):2678-2689. [62] 赵佳丽,陈秋红,李红娜,等.神经病理性疼痛的药物治疗进展[J].药学实践杂志,2016, 34(4):309-312. [63] 李旭,申乐,许力,等.突触后致密物蛋白质95基因沉默及对神经病理性疼痛大鼠疼痛行为的干预[J].协和医学杂志,2011,2(4):343-349. [64] CORTES-ALTAMIRANO JL, OLMOS-HERNANDEZ A, JAIME HB, et al. Review: 5-HT1, 5-HT2, 5-HT3 and 5-HT7 Receptors and their Role in the Modulation of Pain Response in the Central Nervous System. Curr Neuropharmacol. 2018;16(2):210-221. [65] HEIJMANS L, MONS MR, JOOSTEN EA. A systematic review on descending serotonergic projections and modulation of spinal nociception in chronic neuropathic pain and after spinal cord stimulation. Mol Pain. 2021;17: 17448069211043965. [66] HIROKI T, SUTO T, OHTA J, et al. Spinal γ-Aminobutyric Acid Interneuron Plasticity Is Involved in the Reduced Analgesic Effects of Morphine on Neuropathic Pain. J Pain. 2022; 23(4):547-557. [67] RESSLER KJ, NEMEROFF CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12 Suppl 1:2-19. [68] MELE T, CARMAN-KRZAN M, JURIC DM. Regulatory role of monoamine neurotransmitters in astrocytic NT-3 synthesis. Int J Dev Neurosci. 2010;28(1):13-19. [69] CHIANG CY, SESSLE BJ, DOSTROVSKY JO. Role of astrocytes in pain. Neurochem Res. 2012;37(11): 2419-2431. [70] DAS SARMA J. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J Neurovirol. 2014; 20(2):122-136. [71] SHI K, ZHANG J, DONG JF, et al. Dissemination of brain inflammation in traumatic brain injury. Cell Mol Immunol. 2019;16(6):523-530. [72] 王振鹏,王开强.星形胶质细胞和小胶质细胞介导的神经病理痛研究进展[J].现代中西医结合杂志,2017,26(10):1134-1137. [73] GORINA R, FONT-NIEVES M, MÁRQUEZ-KISINOUSKY L, et al. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242-255. [74] KLEGERIS A, GIASSON BI, ZHANG H, et al. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006;20(12):2000-2008. [75] KAWASAKI Y, ZHANG L, CHENG JK, et al. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28(20):5189-5194. [76] HOULDSWORTH A. Role of oxidative stress in neurodegenerative disorders: a review of reactive oxygen species and prevention by antioxidants. Brain Commun. 2024;6(1):fcad356. [77] SINGH A, KUKRETI R, SASO L, et al. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules. 2019;24(8):1583. [78] NIEDZIELSKA E, SMAGA I, GAWLIK M, et al. Oxidative Stress in Neurodegenerative Diseases. Mol Neurobiol. 2016;53(6):4094-4125. [79] KONG J, FAN R, ZHANG Y, et al. Oxidative stress in the brain-lung crosstalk: cellular and molecular perspectives. Front Aging Neurosci. 2024;16:1389454. [80] AYALA A, MUÑOZ MF, ARGÜELLES S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. [81] VALKO M, LEIBFRITZ D, MONCOL J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44-84. [82] RISTOW M, ZARSE K, OBERBACH A, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106(21):8665-8670. [83] DA SILVA LF, DESANTANA JM, SLUKA KA. Activation of NMDA receptors in the brainstem, rostral ventromedial medulla, and nucleus reticularis gigantocellularis mediates mechanical hyperalgesia produced by repeated intramuscular injections of acidic saline in rats. J Pain. 2010; 11(4):378-387. [84] HU C, HE M, CHEN M, et al. Amelioration of Neuropathic Pain and Attenuation of Neuroinflammation Responses by Tetrahydropalmatine Through the p38MAPK/NF-κB/iNOS Signaling Pathways in Animal and Cellular Models. Inflammation. 2022;45(2):891-903. [85] 吕虎,陈辉,熊源长.活性氧在神经病理性疼痛中的作用机制研究进展[J].中国疼痛医学杂志,2013,19(3):130-133+138. [86] BARRÈS R, YAN J, EGAN B, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15(3):405-411. [87] HAMED NJ, GHARAKHANLOU R, PEERI M. The Effect of Endurance Training on Fibronectin Gene Expression of the Sciatic Nerve in Diabetic Rats. Acta Medica Iranica. 2020;58(2):50. [88] JIN H, CHIOU TT, SERWANSKI DR, et al. Ring finger protein 34 (RNF34) interacts with and promotes γ-aminobutyric acid type-A receptor degradation via ubiquitination of the γ2 subunit. J Biol Chem. 2014;289(42):29420-29436. [89] OHSAWA M, BRAILOIU GC, SHIRAKI M, et al. Modulation of nociceptive transmission by pituitary adenylate cyclase activating polypeptide in the spinal cord of the mouse. Pain. 2002;100(1-2):27-34. [90] MABUCHI T, SHINTANI N, MATSUMURA S, et al. Pituitary adenylate cyclase-activating polypeptide is required for the development of spinal sensitization and induction of neuropathic pain. J Neurosci. 2004;24(33):7283-7291. [91] SLUKA KA, O’DONNELL JM, DANIELSON J, et al. Regular physical activity prevents development of chronic pain and activation of central neurons. J Appl Physiol (1985). 2013;114(6):725-733. |
| [1] | Zhang Qingtong, Chen Leqin, Liu Chang, Chen Yuting, Guo Ruiwu. Neuromechanism of the endocannabinoid system in regulating exercise motivation [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(在线): 1-11. |
| [2] | Liu Jinlong, Abuduwupuer·Haibier, Bai Zhen, Su Danyang, Miao Xin, Li Fei, Yang Xiaopeng. Efficacy of different nonsurgical treatments for adolescent idiopathic scoliosis: a systematic review and network meta-analysis [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(9): 2370-2379. |
| [3] | Cai Ziming, Yu Qinghe, Ma Pengfei, Zhang Xin, Zhou Longqian, Zhang Chongyang, Lin Wenping. Heme oxygenase-1 alleviates lipopolysaccharide-induced inflammatory response in nucleus pulposus mesenchymal stem cells [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1624-1631. |
| [4] | He Jiale, Huang Xi, Dong Hongfei, Chen Lang, Zhong Fangyu, Li Xianhui. Acellular dermal matrix combined with adipose-derived stem cell exosomes promotes burn wound healing [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1699-1710. |
| [5] | Xia Linfeng, Wang Lu, Long Qianfa, Tang Rongwu, Luo Haodong, Tang Yi, Zhong Jun, Liu Yang. Human umbilical cord mesenchymal stem cell-derived exosomes alleviate blood-brain barrier damage in mice with septic encephalopathy [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1711-1719. |
| [6] | Cui Lianxu, Li Haomin, Xu Junrong, Tan Baodong, Lu Dahong, Peng Siwei, Wang Jinhui. Effect of umbilical cord mesenchymal stem cell conditioned medium on tissue repair after traumatic craniocerebral injury in miniature pigs [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1730-1735. |
| [7] | Pan Dong, Yang Jialing, Tian Wei, Wang Dongji, Zhu Zheng, Ma Wenchao, Liu Na, Fu Changxi. Resistance exercise activates skeletal muscle satellite cells in aged rats: role of adiponectin receptor 1 pathway [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1736-1746. |
| [8] | Cao Yong, Teng Hongliang, Tai Pengfei, Li Junda, Zhu Tengqi, Li Zhaojin. Interactions between cytokines and satellite cells in muscle regeneration [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(7): 1808-1817. |
| [9] | Hou Chaowen, Li Zhaojin, Kong Jianda, Zhang Shuli. Main physiological changes in skeletal muscle aging and the multimechanism regulatory role of exercise [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1464-1475. |
| [10] | Sun Yaotian, Xu Kai, Wang Peiyun. Potential mechanisms by which exercise regulates iron metabolism in immune inflammatory diseases [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1486-1498. |
| [11] | You Huijuan, Wu Shuzhen, Rong Rong, Chen Liyuan, Zhao Yuqing, Wang Qinglu, Ou Xiaowei, Yang Fengying. Macrophage autophagy in lung diseases: two-sided effects [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1516-1526. |
| [12] | Yin Yongcheng, Zhao Xiangrui, Yang Zhijie, Li Zheng, Li Fang, Ning Bin. Effect and mechanism of peroxiredoxin 1 in microglial inflammation after spinal cord injury [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1106-1113. |
| [13] | Zhang Di, Zhao Jun, Ma Guangyue, Sun Hui, Jiang Rong. Mechanism of depression-like behavior in chronic social defeat stress mice based on high-throughput sequencing [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1139-1146. |
| [14] | Li Haojing, Wang Xin, Song Chenglin, Zhang Shengnan, Chen Yunxin. Therapeutic efficacy of extracorporeal shock wave therapy in the upper trapezius muscle area combined with exercise control training in patients with chronic non-specific neck pain [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1162-1170. |
| [15] | Liu Yu, Lei Senlin, Zhou Jintao, Liu Hui, Li Xianhui. Mechanisms by which aerobic and resistance exercises improve obesity-related cognitive impairment [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1171-1183. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||