Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (10): 2536-2549.doi: 10.12307/2026.644
Previous Articles Next Articles
Glial-neuronal interactions in basal ganglia neurodegenerative diseases: regulatory mechanisms and potential therapeutic targets
Li Minghui1, 2, Zhang Yingbi1, 2, Zhang Xiaorui1, Yin Jihong1, 3, Wang Peng1
- 1Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China; 2Reproductive Center of Beihua University Affiliated Hospital, Jilin 132013, Jilin Province, China; 3The Second Intensive Care Unit, Jilin Central Hospital, Jilin 132013, Jilin Province, China
-
Received:2025-05-21Accepted:2025-06-20Online:2026-04-08Published:2025-08-29 -
Contact:Wang Peng, PhD, Professor, Master’s supervisor, Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China -
About author:Li Minghui, MS candidate, Department of Human Anatomy, School of Basic Medical Sciences, Beihua University, Jilin 132013, Jilin Province, China; Reproductive Center of Beihua University Affiliated Hospital, Jilin 132013, Jilin Province, China -
Supported by:Science and Technology Development Program of Jilin Provincial Department of Science and Technology, No. 20240402007GH (to WP)
CLC Number:
Cite this article
Li Minghui, Zhang Yingbi, Zhang Xiaorui, Yin Jihong, Wang Peng. Glial-neuronal interactions in basal ganglia neurodegenerative diseases: regulatory mechanisms and potential therapeutic targets[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(10): 2536-2549.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
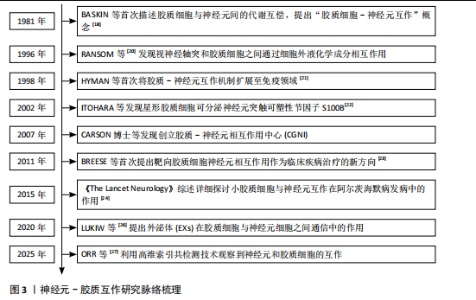
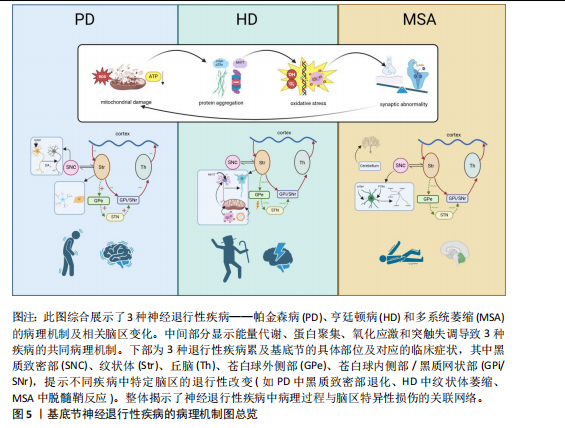
2.1 神经元-胶质互作概述 长期以来,神经元一直是神经系统细胞研究的重心。随着分子遗传学、细胞和突触研究的新突破,人们对中枢神经系统结构和功能的理解也有了新的认识。胶质细胞逐渐从“神经元的支持细胞”转变为神经环路动态调控的关键参与者。胶质-神经元互作的研究始于20世纪末。1981年BASKIN等在神经退行性疾病的早期病理研究中发现,基底节与皮质区域的神经元特异性蛋白异常积累与胶质纤维酸性蛋白水平升高呈现同步性特征,这一现象在约瑟夫病、亨廷顿病及多系统萎缩症中普遍存在。该发现提示胶质细胞增生并非单纯继发于神经元损伤的被动反应,而可能通过增强代谢支持参与疾病代偿机制。基于此,研究首次系统性阐释了胶质细胞与神经元间通过能量底物供给、代谢废物清除等途径形成的双向调控模式,并将这一动态平衡定义为胶质-神经元互作(Glial-neuronal interactions)的核心内涵[18]。随后该领域研究广泛展开[19],RANSOM等[20]发现视神经轴突和胶质细胞之间通过胶质细胞或神经元刺激引起细胞外液化学成分变化,包括离子(如K+或H+)或小分子(如谷氨酸或ATP),进一步明确了胶质细胞与神经元间互作过程中涉及的物质成分。XIA等[21]应用免疫组化方法分析了趋化因子CCR3、CCR5及其配体在正常大脑和阿尔茨海默病大脑中的表达,结果发现CCR3+或CCR5+反应性小胶质细胞、巨噬细胞炎症蛋白反应性星形胶质细胞与淀粉样蛋白沉积密切相关,同时在神经元中可检测到它们的配体。这提示趋化因子等免疫机制在胶质-胶质和胶质-神经元相互作用中发挥作用,并影响阿尔茨海默病的进展,这一研究结果将胶质-神经元互作机制扩展至免疫领域及神经炎症。NISHIYAMA等[22]证明了星形胶质细胞合成的钙结合蛋白S100B能够调节长时程突触可塑性。突变小鼠在水迷宫测试中表现出空间记忆增强,在情境恐惧条件反射中表现出增强的恐惧记忆,提出S100B是神经元突触可塑性的胶质调节因子,胶质细胞的免突触修剪作用被揭示。随着单细胞测序和类器官模型等技术的发展,胶质-神经元互作在基底节退行性疾病中的分子作用机制也逐渐被揭示。KNAPP等[23]通过大鼠中央杏仁核微量注射肿瘤坏死因子α等体内实验证实,肿瘤坏死因子α增加了中央杏仁核神经元的放电频率和γ-氨基丁酸释放。阻断中枢神经系统中潜在的胶质细胞-神经元相互作用可能为改变压力诱导的情绪状态提供解决方法,首次提出靶向胶质细胞-神经元相互作用为临床疾病治疗的新方向。小胶质细胞参与阿尔茨海默病发病的部分,对小胶质细胞与神经元间的互作对于推动疾病发展以及作为疾病靶点的可能性进行了详细的探讨[24]。ZHOU等[25]提出利用光激活腺苷酸环化酶在体内增加星形胶质细胞环磷酸腺苷可以诱导突触可塑性并调节记忆,并发现其作用机制是通过星形胶质细胞-神经元乳酸穿梭介导的,通过动物实验揭示了星形胶质细胞环磷酸腺苷在大脑功能中的作用。LUKIW等[26]提出近年来研究发现,胶质细胞通过分泌外泌体及微囊泡等纳米级膜泡结构,在神经退行性微环境中建立新型细胞间通讯模式;星形胶质细胞与活化小胶质细胞释放的膜泡可选择性携带miRNA-34a、miRNA-146a等调控因子,通过血脑屏障渗透或突触间隙扩散实现跨细胞转运。这类miRNA分子在靶神经元内可特异性调控炎症相关通路(如核因子κB)及氧化应激反应,进而诱导tau蛋白异常磷酸化或β-淀粉样蛋白沉积等病理改变,这种基于膜泡载体的信息传递机制,揭示了神经胶质网络通过表观遗传调控参与退行性病变进程的重要途径。2024年,ORR等[27]利用高维索引共检测技术,研究了继发进展型多发性硬化和原发进展型多发性硬化中多发性硬化病变的细胞微环境,分析了2个独立队列中多发性硬化病变和外观正常的白质内的免疫细胞、胶质细胞、神经元和内皮细胞的相互作用,结果显示,多发性硬化病变中EB病毒标志物EB病毒核抗原1和潜伏性膜蛋白1显著富集,表明EB病毒在多发性硬化中直接参与了神经元和胶质细胞的病变。据此,抗病毒药物和脑渗透性免疫调节剂等潜在治疗靶点被提出可能有助于解决EB病毒对多发性硬化进展的影响。综上可知,整脑功能不仅依赖于神经元的独立功能单位,胶质细胞如星形胶质细胞、小胶质细胞和形成髓鞘的少突胶质细胞的角色也不可或缺,这3种胶质细胞与神经元紧密关联,并以多种方式调节其特定功能[28-30],该领域研究的时间脉络图见图3。 2.2 胶质细胞-神经元互作在基底节退行性疾病中的作用机制 胶质细胞在神经炎症过程中扮演了“双刃剑”的角色:在正常生理范畴,它们发挥正性作用,参与突触的可塑性,促进神经环路的发育,对维持中枢神经系统正常的神经元功能至关重要[13]。星形胶质细胞及小胶质细胞可与神经元突触之间形成较为密切的直接接触[31],还可通过释放细胞因子和可溶性因子,如脑源性神经营养因子、谷氨酸、肿瘤坏死因子α、白细胞介素1β、甘氨酸和L-丝氨酸等,直接作用于神经元,影响基础神经传递和突触可塑性[32]。星形胶质细胞除了谷氨酸外,还可以分泌其他递质,如γ-氨基丁酸、ATP和D-丝氨酸等,通过与神经元的相应受体结合,调节突触传递和可塑性[33]。这些过程使大脑依赖于不间断的能量供应,H?SLI等[34]发现星形胶质细胞与脉管系统相连,维持糖酵解功能,维持自身需求并调节对神经元和突触的能量,当能量供应失败时,脉管系统衰竭或星形胶质细胞病理学,大脑代谢和随后的神经传递处于危险之中。另外神经元释放的谷氨酸被星形胶质细胞摄取,转化为谷氨酰胺后作用神经元,可调控突触活动能量需求。因此支持代谢功能至关重要[35]。激活的M1小胶质细胞能量消耗依赖糖酵解,过程中产生大量乳酸和活性氧,这些产物又加剧神经元氧化应激[36]。神经元-胶质互作通过动态能量供应、代谢废物清除和线粒体支持维持脑能量稳态,其失衡是神经退行性疾病的核心病理环节,可见神经元与胶质细胞之间以及胶质细胞之间的双向通信在大脑功能中起着关键作用,见图4。最近的研究表明,异常的神经元-胶质相互作用存在于大多数已知的中枢神经病理状况,包括帕金森病、亨廷顿病、多系统萎缩以及神经发育障碍如自闭症谱系障碍和抑郁症等[37]。 2.2.1 胶质-神经元互作在帕金森病中的作用机制 帕金森病是仅次于阿尔茨海默病的第2大常见神经退行性疾病,其临床症状包括运动症状(如"
"
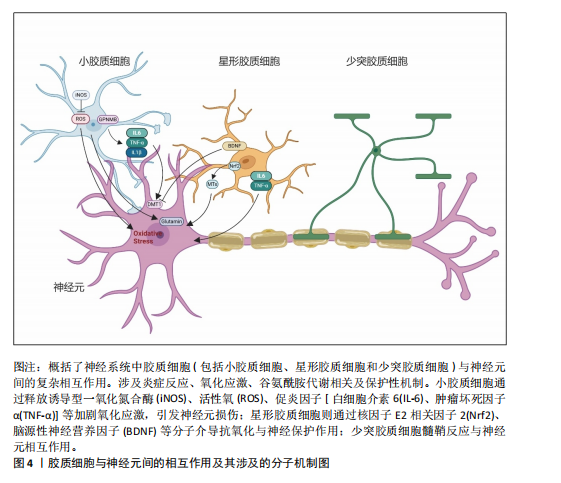
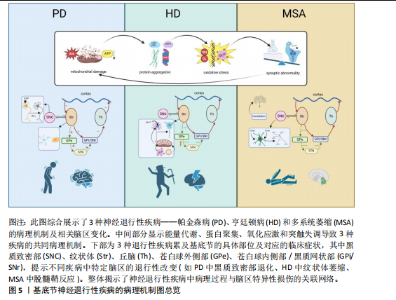
运动迟缓、静止性震颤、僵硬和姿势不稳)和非运动症状(如嗅觉减退、便秘、睡眠障碍和抑郁)。在欧洲,帕金森病的患病率和发病率分别为(108-257)/10万和(11-19)/10万[38]。截至2021年国内帕金森病的发病率、患病率和死亡率分别为35.73/10万、356.85/10万、6.47/10万[39]。帕金森病的主要病理改变特征是黑质致密部多巴胺能神经元的丢失以及路易体中错误折叠的α-突触核蛋白累积。α-突触核蛋白在体内以无毒且可溶的单体形式存在,但其聚集会导致毒性功能的获得[40]。研究表明多种遗传和环境因素调节α-突触核蛋白单体向聚集体转化,这一过程涉及多种细胞参与[41]。帕金森病的病因尚不完全清楚,但研究表明遗传因素(PARK等基因突变)以及污染物、农药、重金属和感染等环境因素可能与患病风险增加相关[42]。 帕金森病中,小胶质细胞的异常激活及其与神经元的动态互作是驱动神经炎症与多巴胺能神经元退变的核心机制。早期病理研究已证实,帕金森病患者黑质区小胶质细胞呈现持续性激活状态,表现为促炎标记物诱导型一氧化氮合酶和吞噬表型标记物CD68的显著上调[43-45]。单核RNA测序进一步揭示了帕金森病相关小胶质细胞的异质性:黑质纹状体通路中,嘌呤能受体(P2RY12)高表达的稳态亚群逐渐分化为热休克蛋白90KDaα(胞浆),类A成员1(HSP90AA1/IL1B)或重组人非转移性行黑色素瘤糖蛋白B主导的激活亚群,提示不同功能状态的小胶质细胞可能通过时空特异性方式参与疾病进程[46]。此外,α-突触核蛋白聚集体不仅直接激活小胶质细胞炎症小体,还可作为趋化信号引导其向受损神经元迁移[47],这一过程伴随白细胞介素1β、肿瘤坏死因子α等细胞因子的释放,并通过外周循环募集单核细胞浸润中枢神经系统[48-52]。 在调控层面,小胶质细胞的激活状态受到多重信号通路的精细平衡。抑制性信号轴包括:①趋化因子C-X3-C-基元配体1/趋化因子C-X3-C-基元受体1(CX3CL1-CX3CR1)信号通路:小胶质细胞表达的趋化因子CX3CR1受体通过结合神经元分泌的CX3CL1配体,抑制其过度激活并维持突触可塑性[53]。动物实验表明,纹状体持续输注CX3CL1可显著减轻6-羟多巴胺模型中的小胶质细胞活化与多巴胺能神经元丢失[54];②CD200-CD200R通路:神经元表面CD200与小胶质细胞CD200R的相互作用,对维持小胶质细胞的静息状态至关重要。临床研究显示,帕金森病患者外周来源的巨噬细胞中CD200R的表达水平显著降低,提示该免疫调节通路可能存在功能缺陷。动物实验中,阻断CD200-CD200R结合(如纹状体注射CD200R抗体)可加重6-羟多巴胺模型大鼠的运动障碍,减少黑质多巴胺能神经元数量,并伴随小胶质细胞异常活化及促炎因子释放。这表明CD200-CD200R系统功能缺陷会解除对小胶质细胞的抑制,加速神经元退行性变[55]。促进性信号轴则以CC趋化因子配体2(CC chemokine ligand 2,CCL2)/CC趋化因子受体2(CC chemokine receptor,CCR2)通路为代表:α-突触核蛋白异常聚集诱导CCL2分泌,通过激活外周单核细胞CCR2受体促进其向中枢神经系统浸润,这一过程与帕金森病患者血清CCL2水平及疾病亚型显著相关[52]。此外,多项帕金森动物模型表明,内源性大麻素系统通过小胶质细胞大麻素受体2的激活发挥神经保护作用,而基质金属蛋白酶2/基质金属蛋白酶9的异常活化可能破坏血脑屏障完整性,形成神经炎症的正反馈环路[56-58]。 在帕金森病的病理进程中,胶质细胞与神经元间交互网络的失衡形成了自我强化的恶性循环:过度激活的小胶质细胞释放白细胞介素6、肿瘤坏死因子α等促炎因子,不仅直接诱导多巴胺能神经元损伤,还会通过抑制星形胶质细胞谷氨酸转运体谷氨酸转运体1的功能,导致突触间隙谷氨酸清除障碍及兴奋性毒性[59-60]。这种神经炎症与神经退行性变的相互促进,提示通过靶向调控关键通路(如增强CX3CL1/CD200抑制性信号或阻断CCL2/基质金属蛋白酶促炎通路)可能重塑胶质-神经元稳态,从而为干预帕金森病的疾病进展提供新策略。 2.2.2 胶质-神经元互作在多系统萎缩中的作用机制 多系统萎缩是一种罕见、进行性且致命的神经退行性疾病,其特征包括自主神经功能障碍、帕金森综合征和小脑性共济失调,其发病率估计为(0.6-0.7)/10万[7]。根据主要症状,多系统萎缩在临床上分为2种亚型:以橄榄桥小脑萎缩引起小脑性共济失调为主的多系统萎缩-C型,以纹状体黑质变性导致帕金森综合征为主的多系统萎缩-P型[61]。多系统萎缩的病理标志是少突胶质细胞胞质中α-突触核蛋白的异常积累,称为胶质细胞质包涵体。从机制上看,多种因素参与了多系统萎缩的发病机制,包括α-突触核蛋白的异常积累、小胶质细胞激活和神经炎症、自噬功能障碍、线粒体功能障碍以及蛋白酶体功能障碍[62]。其中小胶质细胞激活和神经炎症也是多系统萎缩的重要特征之一[63-65]。研究表明,Toll样受体4(Toll-like receptor 4,TLR4)介导的小胶质细胞对α-突触核蛋白的吞噬作用,诱导核因子κB易位、活性氧生成以及促炎细胞因子(如肿瘤坏死因子α)的释放[66]。α-突触核蛋白激活小胶质细胞还依赖于TLR1/2信号通路,这表明TLRs在α-突触核蛋白诱导的小胶质细胞促炎反应和活性氧释放中具有调节作用[67]。最新研究在多系统萎缩患者的脑组织中发现一些含有α-突触核蛋白的小胶质细胞远离含有胶质细胞质内含物的少突胶质细胞,因此推测这些小胶质细胞在摄取α-突触核蛋白后并未就地降解这些错误折叠的蛋白质,而是作为载体在多系统萎缩中传播α-突触核蛋白[68],这提示特异性抑制运输α-突触核蛋白的胶质细胞迁移可成为潜在的治疗措施。此外,在多系统萎缩患者的死后脑组织和病毒介导的多系统萎缩小鼠模型中,均观察到T细胞浸润伴炎性小胶质细胞增生,纹状体组织中Th1 T细胞的比例和干扰素γ细胞因子的水平显著增加,这证实少突胶质细胞中的α-突触核蛋白可诱导小胶质细胞激活,触发CD4+ T细胞浸润。CD4+ T细胞浸润中枢神经系统后,其T细胞受体与小胶质细胞表面上调的组织相容性复合体Ⅱ结合,促进CD4 T细胞分化为Th1 T细胞并分泌干扰素γ,这一途径导致少突胶质细胞功能障碍以及纹状体和胼胝体脱髓鞘,产生多系统萎缩主要症状[69]。可见T细胞浸润中枢神经系统及其与小胶质细胞的相互作用是多系统萎缩疾病发病机制的关键环节。 2.2.3 胶质细胞-神经元互作在亨廷顿病中的作用机制 亨廷顿病是一种遗传性神经退行性疾病,表现为不自主的舞蹈样动作以及认知和行为障碍。亨廷顿病是一种单基因常染色体显性遗传病,由亨廷顿蛋白基因中CAG三核苷酸重复序列的扩展引起。该突变导致亨廷顿蛋白中出现异常长的多聚谷氨酰胺扩展,使蛋白质易于错误折叠[70]。发达国家人群中亨廷顿病的发病率为(4-10)/10万,平均发病年龄为40岁[71]。致病性突变亨廷顿蛋白在大脑的多种神经元中表达,异常聚集的亨廷顿蛋白通常以包涵体的形式存在于细胞质或细胞核中,导致纹状体中的中型多棘神经元选择性丢失以及尾状核和壳核萎缩[72]。目前普遍认为N端亨廷顿蛋白片段在亨廷顿病发病机制中起重要作用,较小的N端亨廷顿蛋白片段比较大的片段表现出更强的神经毒性[73]。 在亨廷顿病患者的纹状体中常发现反应性胶质增生,包括反应性纤维性星形胶质细胞增生和反应性小胶质细胞增生[74]。在皮质-纹状体脑切片和原代神经元培养模型中,神经元中亨廷顿蛋白的表达导致小胶质细胞激活。这些反应性小胶质细胞倾向于聚集在异常神经突起周围,但并不直接导致神经元退化[75]。亨廷顿病中小胶质细胞的激活涉及多种信号通路,包括核因子κB通路等,小胶质细胞检测外部刺激激活后,TLRs触发一系列信号通路,导致免疫反应的激活。TLRs信号通过细胞内适配髓样分化因子88(myeloid differentiation factor 88,MyD88)因子激活下游核因子κB信号级联反应,从而增加促炎细胞因子的产生。研究表明,可溶性亨廷顿蛋白激活IκB激酶,触发核因子κB信号通路,并导致促炎细胞因子基因表达的增加[76]。通过siRNA降低亨廷顿蛋白水平改善了核因子κB转录失调并减少了亨廷顿病中促炎细胞因子的产生[77]。这些发现表明小胶质细胞动态状态的变化可能导致其在亨廷顿病中的神经支持功能受损,见图5。 2.3 胶质-神经元互作技术前沿 胶质神经元互作技术是研究神经系统功能、发育和疾病的重要工具,其中类器官模型和单细胞测序技术的结合极大地推动了这一领域的发展。"
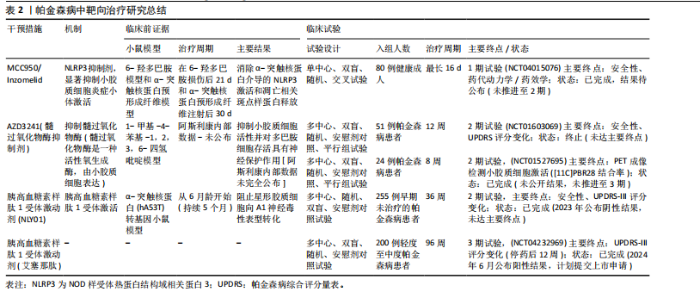
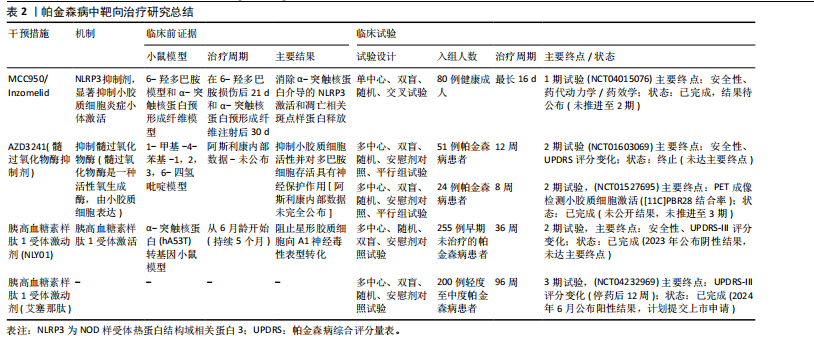
2.3.1 类器官模型的贡献 脑类器官技术是通过模拟体内微环境,揭示胶质细胞对神经发育以及细胞间复杂的调控关系。ZHENG等[78]将星形胶质细胞的条件培养基加入到人源多能干细胞衍生的二维和三维神经培养系统中,发现星形胶质细胞分泌的因子可以加速神经元的分化,增强神经元网络的电生理活性,还能通过脂滴介导的应激保护机制,促进脑类器官的功能成熟。PARK等[79]将人诱导多能干细胞衍生的原始样巨噬细胞与脑类器官共培养,在该模型中,原始样巨噬细胞分化为小胶质样细胞,并通过脂肪分化相关蛋白脂滴介导胆固醇及其酯的输出,促进神经祖细胞的脂质摄取,进而调控神经发生过程,抑制细胞增殖并增强轴突形成。这一研究填补了脑类器官中小胶质细胞功能研究的空白,并在小鼠和人胚胎脑中验证了该脂质代谢互作机制的保守性,提示其在神经发生中的普适性。CAKIR等[80]在人胚胎干细胞来源的皮质类器官中过表达髓系转录因子PU.1,构建了含有功能性小胶质细胞的类器官模型,他们发现这些小胶质细胞可以激活补体/趋化因子系统,减轻β-淀粉样蛋白诱导的神经元损伤,抑制凋亡和铁死亡相关基因的表达;此外,他们还利用混合成簇规律间隔的短回文重复序列干扰(clustered regularly interspaced short palindromic repeat interference,CRISPRi)和单细胞转录组技术解析了阿尔茨海默病风险基因的功能,为研究小胶质细胞在神经退行性疾病中的作用提供了新的研究平台。 2.3.2 单细胞测序的贡献 FLECK等[81]通过整合了人神经类器官的单细胞多组学数据,并利用自主开发的Pando框架构建了全局基因调控网络,研究发现,转录因子GLI3通过调控HES4/5,影响背腹模式和神经节隆起的多样化,从而决定了端脑的命运。结合混合遗传扰动实验,进一步验证了转录因子对细胞命运和状态的调控层级,为利用类器官模型解析人类大脑发育的基因调控机制提供了系统性的研究范式。WU等[82]首次发现周围神经系统中存在一种与中枢小胶质细胞具有相似转录组和表观遗传特征的巨噬细胞群(周围神经系统小胶质样细胞),这些细胞通过包裹神经元胞体,调控发育中的胞体增大和轴突生长;系统发育分析表明,这种细胞在脊椎动物中的存续与神经元胞体大小(而非进化距离)密切相关,尤其在大胞体神经元物种中更为常见。这一发现确立了中枢神经系统小胶质细胞的周围神经系统同源细胞,并揭示了它们在神经元形态进化中的调控作用。 2.3.3 类器官与单细胞测序的联合应用 类器官与单细胞测序的联合应用正在为精准医学的发展提供新的思路。VICTOR等[83]利用CRISPR编辑的诱导多能干细胞模型,发现APOE4等位基因会导致小胶质细胞脂质异常蓄积,增强脂肪生成程序,削弱其对神经元活动的响应能力,同时加剧促炎信号传导,最终导致神经元网络协调性受损,这表明阿尔茨海默病患者中神经元功能障碍可能源于小胶质细胞脂质稳态失衡引发的非细胞自主性调控紊乱。LENG等[84]则结合CRISPR干扰筛选和单细胞转录组学技术,揭示了白细胞介素6/干扰素自分泌-旁分泌信号通过STAT3双向调控炎性星形胶质细胞的反应性。这种反应性在阿尔茨海默病等人类脑疾病及小鼠模型中异常表达,为靶向调节神经炎症反应提供了分子机制和治疗策略。 2.4 靶向胶质细胞-神经元互作的治疗策略 胶质细胞的激活及其介导的炎症反应是神经退行性疾病发展中的关键因素,因此理解胶质细胞激活的复杂性和失衡可能为包括帕金森病在内的神经退行性疾病的治疗干预提供新思路。 2.4.1 帕金森病相关治疗策略 调节或修饰小胶质细胞受体Toll样受体和大麻素受体2为帕金森病治疗提供了新的药理学方法。通过调节Toll样受体2及其下游信号成分蛋白髓样分化(MyD88)、肿瘤坏死因子受体连接因子6和转化生长因子激酶1可对1-甲基-4-苯基-1,2,3,6-四氢吡啶诱导的帕金森病模型鼠具有保护作用[85]。大麻素受体2的天然激动剂β-石竹烯和选择性大麻素受体2受体激动剂JWH133均在模型小鼠中通过调节小胶质细胞激活和抑制促炎细胞因子的表达保护神经元[86]。这些均表明大麻素受体2受体激动剂可能作为帕金森病的潜在治疗靶点。此外,NLRP3抑制剂MCC950显著抑制了炎症小体激活、α-突触核蛋白聚集体的积累以及黑质纹状体多巴胺能神经元的退化,改善帕金森病模型小鼠的运动功能[87]。研究表明,维生素D通过抑制小胶质细胞激活和促进M2极化保护多巴胺能神经元免受炎症和氧化应激的损害,同时增加M2小胶质细胞标志物CD163、CD204和CD206的表达[88]。过氧化物酶体增殖物激活受体γ激动剂罗格列酮通过调节小胶质细胞极化对多巴胺能神经元发挥神经保护作用。罗格列酮减少促炎细胞因子肿瘤坏死因子α和白细胞介素1β,增加抗炎细胞因子转化生长因子β和白细胞介素10,诱导M2极化,并缓解黑质致密部神经元的多巴胺能神经元变性[89]。北美临床试验注册方案已通过的帕金森病临床试验中的小胶质细胞靶向疗法中,NLRP3抑制剂Inzomelid的1期临床试验招募已于2020年完成(NCT04015076),但结果尚未公布。目前暂无美国食品药品监督管理局审批记录,但2019年获得针对克罗恩病的孤儿药资格。美国国立卫生院资助多项与NLRP3相关炎症小体的研究,其中由约翰霍普金斯大学启动的$3.1 million(2018–2023年)资助项目探讨了NLRP3在帕金森病中的作用(R01NS099876),并发现NLRP3抑制剂可减少α-突触核蛋白诱导的神经元死亡[90]。髓过氧化物酶是小胶质细胞等吞噬细胞产生活性氧的关键酶,也是许多疾病中炎症过程的介质[91]。因此,抑制髓过氧化物酶是减少帕金森病神经炎症的一个有吸引力的靶点。选择性不可逆髓过氧化物酶抑制剂AZD3241在帕金森病患者中通过PET成像评估减少了小胶质细胞激活,并且耐受性良好[92]。美国国立卫生院资助了多项与髓过氧化物酶在神经退行性疾病中作用相关的学术研究,其中由宾夕法尼亚大学启动的$2.3 million(2013–2018年)资助项目“髓过氧化物酶与α-突触核蛋白病理的相互作用(R01NS078507)”发现,髓过氧化物酶介导α-突触核蛋白硝基化,并加速其聚集。2023年美国食品药品监督管理局审批通过Cinpanemab(抗α-突触核蛋白单抗),可以靶向清除神经元或胶质细胞内的α-突触核蛋白[93]。胰高血糖素样肽1受体激动剂通常用于治疗2型糖尿病,胰高血糖素样肽1受体激动剂NLY01在hA53T转基因小鼠中直接阻止小胶质细胞介导的星形胶质细胞向A1神经毒性表型的转化[94]。胰高血糖素样肽1类似物司美格鲁肽和利拉鲁肽在慢性1-甲基-4-苯基-1,2,3,6-四氢吡啶诱导的帕金森病小鼠模型中减少了小胶质细胞激活、α-突触核蛋白积累和多巴胺能神经元的丢失,并改善了运动功能障碍[95]。 2021年美国食品药品监督管理局授予其针对帕金森病的快速通道资格,2022年授予其针对帕金森病的孤儿药资格。美国国立卫生研究院资助了多项与胰高血糖素样肽1受体在神经退行性疾病中作用相关的研究,其中由约翰霍普金斯大学启动了$3.2 million (2018-2023年)资助项目“胰高血糖素样肽1受体激活对α-突触核蛋白病理的影响(R01NS099876)”,胰高血糖素样肽1激动剂可减少α-突触核蛋白聚集并改善小鼠运动功能[96]。艾塞那肽是研究最广泛的胰高血糖素样肽1受体激动剂之一,可改善帕金森病患者运动症状,耐受性良好,其效果在给药后可持续12个月[97]。见表2。 2.4.2 多系统萎缩相关治疗策略 在多系统萎缩小鼠模型的早期应用髓过氧化物酶抑制剂,可抑制小胶质细胞激活和神经炎症,减少α-突触核蛋白的胞内聚集,缓解神经元丢失,改善了运动障碍[98]。但在疾病晚期严重多系统萎缩样神经病理学出现以后给予髓过氧化物酶抑制剂进行治疗观察,虽然可检测到小胶质细胞激活减少和α-突触核蛋白的累积,但神经元变性并未得以控制、运动障碍也并无明显改善。这一结果提示抑制小胶质细胞激活治疗时机选择的重要性,"
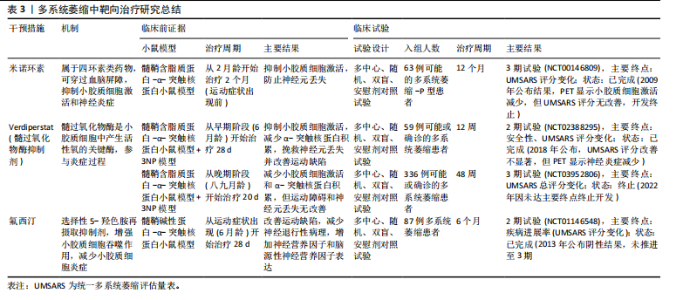
尽早干预小胶质细胞激活是控制疾病进展的重要条件[99]。北美临床试验注册方案已通过:Verdiperstat(髓过氧化物酶抑制剂)已在约250名健康志愿者和患者中进行了1期和2期临床试验。针对多系统萎缩患者的2期试验结果显示,髓过氧化物酶抑制剂治疗12周后,临床评分有所改善,PET成像显示神经炎症减少(NCT02388295)。正在进行中的3期研究以评估Verdiperstat(BHV-3241)治疗48周对多系统萎缩患者的疗效和安全性,其中使用统一多系统萎缩评估量表(Unified Multiple System Atrophy Rating Scale,UMSARS)评分评估verdiperstat的疗效(NCT03952806)。有些靶点抑制在动物模型中虽然效果较好,但临床试验失败。2018年美国食品药品监督管理局针对多系统萎缩适应证授予孤儿药资格,2019年授予快速通道资格,但因Ⅱ/Ⅲ期试验失败,Biohaven终止开发,未向美国食品药品监督管理局提交新药申请。美国国立卫生院资助了多项与髓过氧化物酶在神经退行性疾病中作用相关的研究:其中包括由梅奥诊所启动$2.5 million(2020–2025年)资助项目“髓过氧化物酶介导的氧化损伤在多系统萎缩中的作用(R01NS112345)”,发现髓过氧化物酶活性与少突胶质细胞质内含物密度正相关[100]。如米诺环素,能够穿过血脑屏障并抑制小胶质细胞激活和促炎细胞因子的产生,在转基因多系统萎缩小鼠模型中应用,抑制小胶质细胞激活并防止黑质致密部多巴胺能神经元和纹状体多巴胺能末梢的丢失[101-102]。其后开展的临床试验以评估米诺环素对63例多系统萎缩-P型患者的疗效(NCT00146809),经过12个月的米诺环素治疗,PET成像显示小胶质细胞虽激活减少,但患者运动功能未有明显改善[103]。美国国立卫生院资助了多项与米诺环素神经保护作用相关的基础研究:其中包括埃默里大学启动$0.4 million(2014-2017年)资助米诺环素对帕金森病模型中小胶质细胞极化的影响(R21NS089667),主要评估米诺环素对α-突触核蛋白炎症反应的调控。再如选择性5-羟色胺再摄取抑制剂氟西汀,显著减少脂多糖诱导的炎细胞因子产生和氧化应激,增强了小胶质细胞的吞噬和自噬功能,改善多系统萎缩模型小鼠的运动障碍并减少神经退行性病理的形成[104]。针对多系统萎缩患者的2期双盲、安慰剂对照临床试验研究发现,多系统萎缩患者的进展与对照组相比无差异(NCT01146548)[105]。见表3。 2.4.3 亨廷顿病相关治疗策略 对于亨廷顿病目前尚无特效疗法,现有治疗主要用于缓解舞蹈症和精神症状。鉴于亨廷顿病中的小胶质细胞激活是由神经元亨廷顿蛋白介导的兴奋毒性或小胶质细胞中亨廷顿蛋白表达引起[106],多种治疗方法均以亨廷顿蛋白为靶点,通过减少突变亨廷顿RNA (Htt RNA)的表达,减轻小胶质细胞激活、缓解炎症过程并提供神经保护作用。北美临床试验注册方案已通过:Pepinemab靶向信号素4D的单克隆抗体,抑制信号素4D介导的神经炎症和胶质细胞活化,促进神经元保护和突触可塑性(NCT02481674)[107]。结果显示:可以减少脑萎缩,改善认知功能并减轻焦虑样行为。2021年针对亨廷顿病获批孤儿药资格2022年获美国食品药品监督管理局快速通道资格。目前2期试验已完成,结果未公布。美国国立卫生院资助了多项与信号素4D机制相关的研究,包括加州大学旧金山分校启动$3.5 million(2018–2023年)资助项目“信号素4D在神经炎症中的作用(R01NS099876)”,结果发现抑制信号素4D可减少小胶质细胞活化并改善阿尔茨海默病模型认知功能。反义寡核苷酸——WVE-120101和WVE-120102,靶向亨廷顿病患者中最常见的单核苷酸多态性,已在人体研究中进行了评估(NCT04617847,NCT04617860),结果显示患者脑脊液中的亨廷顿蛋白水平无显著变化。靶向亨廷顿蛋白的反义寡核苷酸 IONIS-HTTRx(也称为ISIS 443139和RG6042)通过RNase H1机制发挥作用,已完成1/2a期临床试"
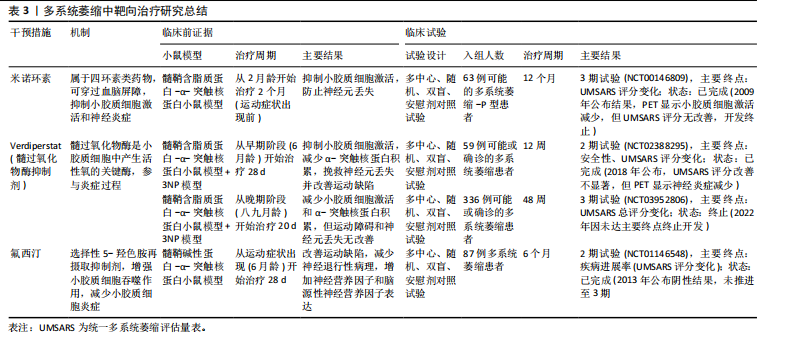
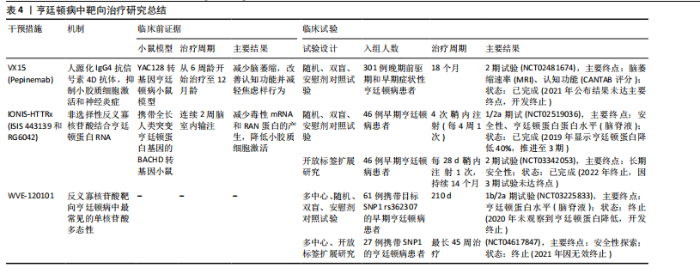
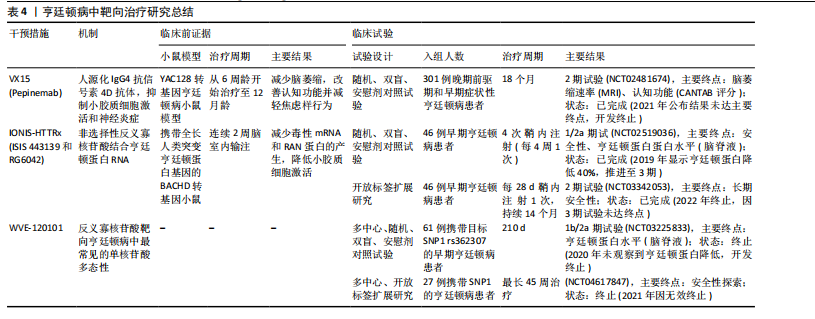
验(NCT02519036,NCT03342053),目前正在进行大型多中心全球疗效研究。阶段性结果显示患者脑脊液中亨廷顿蛋白浓度随给药剂量增加有明显降低,尤其高剂量下的亨廷顿蛋白减少约40%,在安全性上未报告显著不良反应,这为亨廷顿病的治疗带来了希望[108]。见表4。 2.4.4 胶质细胞靶向治疗的挑战 胶质细胞(如小胶质细胞、星形胶质细胞)靶向治疗在神经退行性疾病中展现出潜力,但其临床应用仍面临多重挑战,主要包括血脑屏障穿透效率不足、脱靶效应引发的非特异性毒性,以及胶质细胞功能异质性导致的精准调控难题。以下结合最新研究进展分述关键问题与解决策略:(1)血脑屏障递送障碍:血脑屏障是由脑微血管内皮细胞、星形胶质细胞、周细胞和基膜组成的,它能挡住98%以上的小分子药物和几乎所有大分子(比如抗体、基因编辑工具)进入大脑。虽然传统脂溶性药物可以被动扩散进去,但大多数胶质细胞靶向治疗还是需要主动转运。近年递送技术创新聚焦以下方向:①纳米载体系统:脂质体、聚合物纳米颗粒通过表面修饰(如转铁蛋白受体抗体)可增强血脑屏障穿透性。例如,LI团队[109]在2023年一项研究开发了载有小干扰RNA(small interfering RNA,siRNA)的聚乳酸-羟基乙酸纳米颗粒,通过靶向低密度脂蛋白受体相关蛋白1,成功将siRNA递送至阿尔茨海默病模型小鼠的小胶质细胞,显著降低促炎因子表达;②聚焦超声联合微泡:聚焦超声与微泡相结合,可以局部和瞬时打开血脑屏障,提高药物局部浓度,为药物通过 血脑屏障 输送到大脑提供了一种潜在的策略。GASCA-SALAS等[110]通过一项前瞻性单臂Ⅰ期临床试验(NCT03608553)证实,MR引导聚焦超声(MRgFUS)联合微泡靶向开放右侧顶枕颞叶皮质血脑屏障在帕金森病伴痴呆患者中具有安全性和可重复性,10次治疗中8次通过钆增强显影成功实现血脑屏障可逆开放且无严重不良反应,尽管淀粉样蛋白负荷与脑代谢未见显著变化,但观察到轻度认知改善,为神经退行性疾病中血脑屏障靶向药物递送策略的临床应用提供了可行性依据。 (2)脱靶效应与细胞特异性调控:胶质细胞在中枢神经系统里分布很广,功能也很复杂,所以靶向治疗的时候一定要避免误伤正常细胞。PROFACI等[111]之前用集落刺激因子1受体抑制剂PLX5622来耗竭小胶质细胞,发现虽然小胶质细胞和脑内皮细胞直接接触,但它对健康状态下血脑屏障的结构完整性、功能维持和基因表达其实并不是必需的。不过,PLX5622处理还是会干扰脑内皮细胞的胆固醇代谢,这说明药物对脑血管系统有脱靶效应,也揭示了小胶质细胞和内皮细胞互作的非依赖性以及药理学干预的潜在非特异性影响。这几年的解决方案主要有以下2个方向:①细胞外囊泡:YAO等[112]通过设计RNA适配体(com)与适体结合蛋白(Com-CD63融合蛋白)的特异性互作机制,将Cas9或腺嘌呤碱基编辑器核糖核蛋白主动富集至细胞外囊泡中,构建了高效、瞬时且可多重编辑的递送系统,该系统支持多基因靶点、跨物种Cas9协同应用及体内活性验证,显著提升基因组编辑效率并降低脱靶风险,为基于细胞外囊泡的CRISPR治疗提供了安全可控的技术路径;②细胞类型特异性启动子:SERRANO等[113]基于人IBA1启动子466 bp片段构建腺相关病毒载体,结合miR124靶序列调控,实现了静息态与反应性小胶质细胞的高特异性靶向(转导细胞中95%为小胶质细胞),为解析小胶质细胞功能及开发其靶向基因治疗提供了高效、精准的递送工具。 虽然胶质细胞靶向治疗在神经疾病干预中很有前景,但目前还是受到血脑屏障递送效率、细胞异质性导致的脱靶风险以及胶质细胞和神经元互作复杂调控网络的限制。不过,最近纳米载体、表观遗传编辑和类器官-芯片模型等技术的突破,为解决这些问题提供了新的思路。未来还需要更深入地研究胶质细胞亚群的功能动态"
| [1] DI TELLA S, ZINZI P, ANZUINO I, et al. Social cognition in basal ganglia pathologies: Theory of Mind in Huntington’s and Parkinson’s diseases. Soc Cogn Affect Neurosci. 2025;20(1):nsaf007. [2] YIN Z, YUAN T, YANG A, et al. Contribution of basal ganglia activity to REM sleep disorder in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2024;95(10):947-955. [3] RICCI C. Neurodegenerative Disease: From Molecular Basis to Therapy, 2nd Edition. Int J Mol Sci. 2024;25(2):967. [4] HUANG Y, ZHANG G, LI S, et al. Innate and adaptive immunity in neurodegenerative disease. Cell Mol Life Sci. 2025;82(1):68. [5] HAN CZ. All Fired Up: Microglial Neuroinflammation in Parkinson’s Disease. Biol Psychiatry. 2025;97(7):669-671. [6] CEPEDA C, TONG XP. Huntington’s disease: From basic science to therapeutics. CNS Neurosci Ther. 2018;24(4):247-249. [7] POEWE W, STANKOVIC I, HALLIDAY G, et al. Multiple system atrophy. Nat Rev Dis Primers. 2022;8(1):56. [8] PRANGE SE, BHAKTA IN, SYSOEVA D, et al. Dendrite injury triggers neuroprotection in Drosophila models of neurodegenerative disease. Sci Rep. 2024;14(1):24766. [9] ADAMCZYK A. Glial-neuronal interactions in neurological disorders: molecular mechanisms and potential points for intervention. Int J Mol Sci. 2023;24(7):6274. [10] 蔡星宇, 杨道锋, 卓雪瑞, 等. 人参皂苷Rg3对脂多糖诱导的胶质细胞-神经元互作损伤模型的保护作用[J]. 四川大学学报(医学版),2024,55(6):1543-1549. [11] 苏一洵, 李晖, 易陈菊. 运用化学遗传学方法鉴定星形胶质细胞调控抑制性突触形成的关键蛋白[J]. 神经损伤与功能重建,2022,17(1):1-3. [12] GARCÍA-CÁCERES C, BALLAND E, PREVOT V, et al. Role of astrocytes, microglia, and tanycytes in brain control of systemic metabolism. Nat Neurosci. 2019;22(1):7-14. [13] ALLEN NJ, LYONS DA. Glia as architects of central nervous system formation and function. Science. 2018;362(6411):181-185. [14] KIRKLEY KS, POPICHAK KA, AFZALI MF, et al. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J Neuroinflammation. 2017;14(1):99. [15] ZHAO XF, HUFFMAN LD, HAFNER H, et al. The injured sciatic nerve atlas (iSNAT), insights into the cellular and molecular basis of neural tissue degeneration and regeneration. Elife. 2022;11:e80881. [16] LANCASTER MA, KNOBLICH JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194): 1247125. [17] IBRAHIM WW, SKALICKA-WOŹNIAK K, BUDZYŃSKA B, et al. NLRP3 inflammasome inhibition and M1-to-M2 microglial polarization shifting via scoparone-inhibited TLR4 axis in ovariectomy/D-galactose Alzheimer’s disease rat model. Int Immunopharmacol. 2023;119:110239. [18] ROSENBERG RN, IVY N, KIRKPATRICK J, et al. Joseph disease and Huntington disease: protein patterns in fibroblasts and brain. Neurology. 1981;31(8):1003-1014. [19] AHMAD SR, ZEYAULLAH M, ALSHAHRANI AM, et al. Deciphering the enigma of neuron-glial interactions in neurological disorders. Front Biosci (Landmark Ed). 2024;29(4):142. [20] RANSOM BR, ORKAND RK. Glial-neuronal interactions in non-synaptic areas of the brain: studies in the optic nerve. Trends Neurosci. 1996;19(8):352-358. [21] XIA MQ, QIN SX, WU LJ, et al. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am J Pathol. 1998;153(1): 31-37. [22] NISHIYAMA H, KNOPFEL T, ENDO S, et al. Glial protein S100B modulates long-term neuronal synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99(6):4037-4042. [23] KNAPP DJ, WHITMAN BA, WILLS TA, et al. Cytokine involvement in stress may depend on corticotrophin releasing factor to sensitize ethanol withdrawal anxiety. Brain Behav Immun. 2011;25(Suppl 1):S146-S154. [24] HENEKA MT, CARSON MJ, EL KHOURY J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388-405. [25] ZHOU Z, OKAMOTO K, ONODERA J, et al. Astrocytic cAMP modulates memory via synaptic plasticity. Proc Natl Acad Sci U S A. 2021;118(3):e2016584118. [26] LUKIW WJ, POGUE AI. Vesicular transport of encapsulated microRNA between glial and neuronal cells. Int J Mol Sci. 2020; 21(14):5078. [27] ORR N, STEINMAN L. Epstein-Barr virus and the immune microenvironment in multiple sclerosis: insights from high-dimensional brain tissue imaging. Proc Natl Acad Sci U S A. 2025;122(11):e2425670122. [28] CORTY MM, FREEMAN MR. Cell biology in neuroscience: architects in neural circuit design: glia control neuron numbers and connectivity. J Cell Biol. 2013;203(3):395-405. [29] ALLEN N, EROGLU C. Cell biology of astrocyte-synapse interactions. Neuron. 2017;96(3):697-708. [30] VEIGA-FERNANDES H, ARTIS D. Neuronal-immune system cross-talk in homeostasis. Science. 2018;359(6383):1465-1466. [31] PEREA G, NAVARRETE M, ARAQUE A. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 2009;32(8):421-431. [32] HARADA K, KAMIYA T, TSUBOI T. Gliotransmitter release from astrocytes: functional, developmental, and pathological implications in the brain. Front Neurosci. 2015;9:499. [33] LAL R, SINGH A, WATTS S, et al. Experimental models of Parkinson’s disease: challenges and opportunities. Eur J Pharmacol. 2024; 980:176819. [34] HÖSLI L, ZUEND M, BREDELL G, et al. Direct vascular contact is a hallmark of cerebral astrocytes. Cell Rep. 2022;39(1):1-9. [35] 熊伟杰, 易陈菊. 胆碱能系统功能障碍对神经退行性疾病的影响[J]. 神经损伤与功能重建,2025,20(3):156-161. [36] 张文滨, 汪永杰, 令垚, 等. 1990-2021年中国帕金森病疾病负担分析和预测[J]. 中华疾病控制杂志,2025,29(1):74-81. [37] ZHENG X, ZHANG H, ZHANG Y, et al. Salidroside ameliorates cerebral ischemic injury and regulates the glutamate metabolism pathway in astrocytes. Front Pharmacol. 2024;15:1472100. [38] SIMANENKOVA A, FUKS O, TIMKINA N, et al. Microglia involvement into acute and chronic brain damage in diabetic rats: impact of GLP-1RA and SGLT-2i. Front Biosci (Landmark Ed). 2024;29(7):265. [39] 曹利华, 赵晖, 贺红娟, 等. 神经元-小胶质细胞串话在抑郁症中的作用及中医药的干预探析[J]. 世界科学技术-中医药现代化,2024,26(12):3086-3096. [40] CARCELES-CORDON M, WEINTRAUB D, CHEN-PLOTKIN AS. Cognitive heterogeneity in Parkinson’s disease: a mechanistic view. Neuron. 2023;111(10):1531-1546. [41] WIRDEFELDT K, ADAMI HO, COLE P, et al. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26(Suppl 1):S1-S58. [42] DOORN KJ, MOORS T, DRUKARCH B, et al. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun. 2014;2:90. [43] 李冬青, 秦晓红, 米立志. α-突触核蛋白的结构生物学研究[J]. 中国生物化学与分子生物学报,2023,39(4):531-544. [44] GERHARD A, PAVESE N, HATTON G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006; 21(2): 404-412. [45] LIU Q, LIU Z, XIE W, et al. Single-cell sequencing of the substantia nigra reveals microglial activation in a model of MPTP. Front Aging Neurosci. 2024;16:1390310. [46] SMAJIĆ S, PRADA-MEDINA CA, LANDOULSI Z, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145(3):964-978. [47] BENNETT JP, KEENEY PM, BROHAWN DG. RNA sequencing reveals small and variable contributions of infectious agents to transcriptomes of postmortem nervous tissues from amyotrophic lateral sclerosis, Alzheimer’s disease and Parkinson’s disease subjects, and increased expression of genes from disease-activated microglia. Front Neurosci. 2019;13:235. [48] SCHRÖDER JB, PAWLOWSKI M, MEYER ZU HÖRSTE G, et al. Immune cell activation in the cerebrospinal fluid of patients with Parkinson’s disease. Front Neurol. 2018;9: 1081. [49] TENTILLIER N, ETZERODT A, OLESEN MN, et al. Anti-inflammatory modulation of microglia via CD163-targeted glucocorticoids protects dopaminergic neurons in the 6-OHDA Parkinson’s disease model. J Neurosci. 2016;36(36):9375-9390. [50] HALL S, JANELIDZE S, SUROVA Y, et al. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci Rep. 2018;8(1):13276. [51] SHI Q, GUTIERREZ RA, BHAT MA. Microglia, Trem2, and neurodegeneration. Neuroscientist. 2025;31(2):159-176. [52] MEUCCI O, FATATIS A, SIMEN AA, et al. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97(14):8075-8080. [53] 李楠, 赵仁清, 王斌. 运动改善小胶质细胞介导的神经炎症在预防帕金森病的作用机制[J].中国生物化学与分子生报, 2024,40(6):779-787. [54] PABON MM, BACHSTETTER AD, HUDSON CE, et al. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:9. [55] ZHANG S, WANG XJ, TIAN LP, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;8:154. [56] PRICE DA, MARTINEZ AA, SEILLIER A, et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur J Neurosci. 2009; 29(11):2177-2186. [57] CHUNG YC, SHIN WH, BAEK JY, et al. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp Mol Med. 2016;48(1):e205. [58] RUAN Z, ZHANG D, HUANG R, et al. Microglial activation damages dopaminergic neurons through MMP-2/-9-mediated increase of blood-brain barrier permeability in a Parkinson’s disease mouse model. Int J Mol Sci. 2022;23(5):2793. [59] HERRERA MORO CHAO D, KIRCHNER MK, PHAM C, et al. Hypothalamic astrocytes control systemic glucose metabolism and energy balance. Cell Metab. 2022;34(10): 1532-1547.e6. [60] BOOTH HDE, WESSELY F, CONNOR-ROBSON N, et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol Dis. 2019;129:56-66. [61] LYOO CH, JEONG Y, RYU YH, et al. Effects of disease duration on the clinical features and brain glucose metabolism in patients with mixed type multiple system atrophy. Brain. 2008;131(Pt 2):438-446. [62] SALVESEN L, WINGE K, BRUDEK T, et al. Neocortical neuronal loss in patients with multiple system atrophy: a stereological study. Cereb Cortex. 2017;27(1):400-410. [63] NYKJAER CH, BRUDEK T, SALVESEN L, et al. Changes in the cell population in brain white matter in multiple system atrophy. Mov Disord. 2017;32(7):1074-1082. [64] 朱琳, 刘军. 多系统萎缩生物标志物的研究进展[J]. 上海交通大学学报(医学版),2020,40(9):1303-1307+1302. [65] KÜBLER D, WÄCHTER T, CABANEL N, et al. Widespread microglial activation in multiple system atrophy. Mov Disord. 2019;34(4):564-568. [66] SHAO QH, YAN WF, ZHANG Z, et al. Nurr1: a vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology. 2019;144:388-399. [67] DANIELE SG, BÉRAUD D, DAVENPORT C, et al. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. 2015;8(376):ra45. [68] VALDINOCCI D, GRANT GD, DICKSON TC, et al. Epothilone D inhibits microglia-mediated spread of alpha-synuclein aggregates. Mol Cell Neurosci. 2018;89:80-94. [69] WILLIAMS GP, MARMION DJ, SCHONHOFF AM, et al. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol. 2020;139(5):855-874. [70] PAN L, FEIGIN A. Huntington’s disease: new frontiers in therapeutics. Curr Neurol Neurosci Rep. 2021;21(3):10. [71] NITTARI G, ROY P, MARTINELLI I, et al. Rodent models of Huntington’s disease: an overview. Biomedicines. 2023;11(12):3331. [72] VAN DER BURG JM, BJÖRKQVIST M, BRUNDIN P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8(8):765-774. [73] 程扬帆, 张斯睿, 商慧芳. 亨廷顿病的诊治进展[J]. 实用医院临床杂志,2024, 21(5):12-16. [74] KRAFT AD, KALTENBACH LS, LO DC, et al. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol Aging. 2012;33(3):621.e17-33. [75] KHOSHNAN A, PATTERSON PH. The role of IκB kinase complex in the neurobiology of Huntington’s disease. Neurobiol Dis. 2011; 43(2):305-311. [76] TRÄGER U, ANDRE R, LAHIRI N, et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(Pt 3):819-833. [77] KOO JH, JANG YC, HWANG DJ, et al. Treadmill exercise produces neuroprotective effects in a murine model of Parkinson’s disease by regulating the TLR2/MyD88/NF-κB signaling pathway. Neuroscience. 2017;356:102-113. [78] ZHENG H, FENG Y, TANG J, et al. Astrocyte-secreted cues promote neural maturation and augment activity in human forebrain organoids. Nat Commun. 2025;16:2845. [79] PARK DS, KOZAKI T, TIWARI SK, et al. iPS-cell-derived microglia promote brain organoid maturation via cholesterol transfer. Nature. 2023;623:397-405. [80] CAKIR B, TANAKA Y, KIRAL FR, et al. Expression of the transcription factor PU.1 induces the generation of microglia-like cells in human cortical organoids. Nat Commun. 2022;13:430. [81] FLECK JS, JANSEN SMJ, WOLLNY D, et al. Inferring and perturbing cell fate regulomes in human brain organoids. Nature. 2023; 621:365-372. [82] WU Z, WANG Y, CHEN WW, et al. Peripheral nervous system microglia-like cells regulate neuronal soma size throughout evolution. Cell. 2025;188:2159-2174.e15. [83] VICTOR MB, LEARY N, LUNA X, et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell. 2022;29:1197-1212.e8. [84] LENG K, ROSE IVL, KIM H, et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat Neurosci. 2022;25: 1528-1542. [85] OJHA S, JAVED H, AZIMULLAH S, et al. β-Caryophyllene, a phytocannabinoid attenuates oxidative stress, neuroinflammation, glial activation, and salvages dopaminergic neurons in a rat model of Parkinson disease. Mol Cell Biochem. 2016;418(1-2):59-70. [86] CALVELLO R, CIANCIULLI A, NICOLARDI G, et al. Vitamin D treatment attenuates neuroinflammation and dopaminergic neurodegeneration in an animal model of Parkinson’s disease, shifting M1 to M2 microglia responses. J Neuroimmune Pharmacol. 2017;12(2):327-339. [87] PISANU A, LECCA D, MULAS G, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTP mouse model of progressive Parkinson’s disease. Neurobiol Dis. 2014;71:280-291. [88] COLL RC, ROBERTSON AA, CHAE JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015; 21(3):248-255. [89] ARATANI Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys. 2018;640:47-52. [90] JUCAITE A, SVENNINGSSON P, RINNE JO, et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain. 2015;138(Pt 9): 2687-2700. [91] PANICKER N, KAM TI, WANG H, et al. Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron. 2022;110(15):2422-2437. [92] ZHANG L, ZHANG L, LI L, et al. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis. 2019;9(1):157-171. [93] AVILES-OLMOS I, DICKSON J, KEFALOPOULOU Z, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis. 2014;4(3):337-344. [94] MCFARTHING K, BUFF S, RAFALOFF G, et al. Parkinson’s disease drug therapies in the clinical trial pipeline: 2023 update. J Parkinsons Dis. 2023;13(4):427-439. [95] KOPP KO, GLOTFELTY EJ, LI Y, et al. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: implications for neurodegenerative disease treatment. Pharmacol Res. 2022;186:106550. [96] STEFANOVA N, GEORGIEVSKA B, ERIKSSON H, et al. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res. 2012;21(4):393-404. [97] ZHAO X, WANG M, WEN Z, et al. GLP-1 receptor agonists: beyond their pancreatic effects. Front Endocrinol (Lausanne). 2021; 12:721135. [98] KAINDLSTORFER C, SOMMER P, GEORGIEVSKA B, et al. Failure of neuroprotection despite microglial suppression by delayed-start myeloperoxidase inhibition in a model of advanced multiple system atrophy: clinical implications. Neurotox Res. 2015;28(3): 185-194. [99] STEFANOVA N, REINDL M, NEUMANN M, et al. Microglial activation mediates neurodegeneration related to oligodendroglial α-synucleinopathy: implications for multiple system atrophy. Mov Disord. 2007;22(15):2196-2203. [100] DODEL R, SPOTTKE A, GERHARD A, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord. 2010;25(1):97-107. [101] RAY RS, KATYAL A. Myeloperoxidase: bridging the gap in neurodegeneration. Neurosci Biobehav Rev. 2016;68:611-620. [102] UBHI K, INGLIS C, MANTE M, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of α-synucleinopathy. Exp Neurol. 2012;234(2):405-416. [103] METZ LM, LI DKB, TRABOULSEE AL, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. 2017;376(22):2122-2133. [104] STEFANOVA N. Translational therapies for multiple system atrophy: bottlenecks and future directions. Auton Neurosci. 2018;211:7-14. [105] YANG HM, YANG S, HUANG SS, et al. Microglial activation in the pathogenesis of Huntington’s disease. Front Aging Neurosci. 2017;9:193. [106] TABRIZI SJ, LEAVITT BR, LANDWEHRMEYER GB, et al. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307-2316. [107] FEIGIN A, EVANS EE, FISHER TL, et al. Pepinemab antibody blockade of SEMA4D in early Huntington’s disease: a randomized, placebo-controlled, phase 2 trial. Nat Med. 2024;30(2):606. [108] TABRIZI SJ, LEAVITT BR, LANDWEHRMEYER GB, et al. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307-2316. [109] LI W, QIU J, LI XL, et al. BBB pathophysiology-independent delivery of siRNA in traumatic brain injury. Sci Adv. 2021;7(1):eabd6889. [110] GASCA-SALAS C, FERNÁNDEZ-RODRÍGUEZ B, PINEDA-PARDO JA, et al. Blood-brain barrier opening with focused ultrasound in Parkinson’s disease dementia. Nat Commun. 2021;12(1):779. [111] PROFACI CP, HARVEY SS, BAJC K, et al. Microglia are not necessary for maintenance of blood-brain barrier properties in health, but PLX5622 alters brain endothelial cholesterol metabolism. Neuron. 2024;112(17):2910-2921.e7. [112] YAO X, LYU P, YOO K, et al. Engineered extracellular vesicles as versatile ribonucleoprotein delivery vehicles for efficient and safe CRISPR genome editing. J Extracell Vesicles. 2021;10(5):e12076. [113] SERRANO C, CANANZI S, SHEN T, et al. Simple and highly specific targeting of resident microglia with adeno-associated virus. Preprint. bioRxiv. 2023; 2023.12.12.571321. |
| [1] | Liu Huan, Zeng Shaopeng, Chen Jun, He Linqian, Yang Ying, Zhang Jing. Aging-related dysregulation of glucose metabolism: crossroads of cancer and neurodegenerative diseases [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(6): 1527-1538. |
| [2] | Leng Xiaoxuan, Zhao Yuxin, Liu Xihua. Effects of different neuromodulatory stimulation modalities on non-motor symptoms in Parkinson’s patients: a network meta-analysis [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(5): 1282-1293. |
| [3] | Li Tingwen, , Zhang Jianhua. Meta-analysis of the effects of aquatic exercise on balance function and motor ability in elderly patients with Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2026, 30(10): 2560-2568. |
| [4] | Lu Ranran, Zhou Xu, Zhang Lijie, Yang Xinling. Dimethyl fumarate alleviates nerve damage in a mouse model of Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(5): 989-994. |
| [5] | Zhang Xin, Guo Baojuan, Xu Huixin, Shen Yuzhen, Yang Xiaofan, Yang Xufang, Chen Pei. Protective effects and mechanisms of 3-N-butylphthalide in Parkinson’s disease cell models [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(30): 6466-6473. |
| [6] | Nan Songhua, Peng Chaojie, Cui Yinglin. Mitochondrial dysfunction and brain aging: a bibliometrics analysis based on the Web of Science Core Collection database [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(26): 5642-5651. |
| [7] | Jiang Qianping, Yang Dan, , , Wan Shilei, Xu Dandan, , , , Cao Lu, , Zhou Jing, , , . Role of O-linked N-acetylglucosamine glycosylation in neurodegenerative diseases and its clinical application prospects [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(26): 5704-5712. |
| [8] | Lei Senlin, Chen Xiaoan, Chen Ping, Wang Zhaofeng. Exercise prevention and treatment of Parkinson’ s disease mediated by brain-derived neurotrophic factor: role and mechanism [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(25): 5454-5468. |
| [9] | Guan Mengya, Ren Binbin, Wang Jingying. Major histocompatibility complex regulates immune responses in Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(25): 5469-5477. |
| [10] | Zhu Liuhui, Zhang Xinyue, Zhu Zhouhai, Yang Xinglong, Guan Ying, Liu Bin. Coiled-coil-helix-coiled-coil-helix domain-containing 2 inhibits apoptosis of Parkinson’ s disease SH-SY5Y cells by promoting mitochondrial autophagy [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(25): 5403-5413. |
| [11] | Zhao Yuxin, Zhang Deqi, Bi Hongyan. Effect of different stimulation modalities of non-invasive brain stimulation on cognitive function in patients with Parkinson’s disease: a network Meta-analysis [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(24): 5212-5223. |
| [12] | Lin Huijie, Huang Yun, Huang Zhihua, Jiang Lixia. Hot topics on exosomes as drug delivery system in central nervous system diseases [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(23): 5013-5021. |
| [13] | Wang Jingying, , Ren Binbin, Ma Suna, Yang Yueyue, , Wu Song, , Guan Mengya, . Mechanism of alpha-synuclein in mitochondrial damage induced by Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(17): 3668-3674. |
| [14] | Liu Ziyu, Geng Dandan, Zhang Runjiao, Liu Qing, Li Yibo, Wang Hongfang, Xie Wenmeng, Wang Wenyu, Hao Jiaxin, Wang Lei. Application of single-cell RNA sequencing technology in Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2025, 29(1): 193-201. |
| [15] | Qi Xue, Li Jiahui, Zhu Yuanfeng, Yu Lu, Wang Peng. Abnormal modification of alpha-synuclein and its mechanism in Parkinson’s disease [J]. Chinese Journal of Tissue Engineering Research, 2024, 28(8): 1301-1306. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||