Chinese Journal of Tissue Engineering Research ›› 2026, Vol. 30 ›› Issue (17): 4446-4456.doi: 10.12307/2026.177
Previous Articles Next Articles
Mechanisms underlying mitophagy, ferroptosis, cuproptosis, and disulfidptosis in Parkinson’s disease
Yu Le1, Nan Songhua1, Shi Zijian1, He Qiqi1, Li Zhenjia1, Cui Yinglin2
- 1Henan University of Chinese Medicine, Zhengzhou 450000, Henan Province, China; 2Henan Provincial Hospital of Traditional Chinese Medicine, Zhengzhou 450000, Henan Province, China
-
Received:2025-06-27Accepted:2025-08-28Online:2026-06-18Published:2025-12-03 -
Contact:Cui Yinglin, Chief physician, Doctoral supervisor, Henan Provincial Hospital of Traditional Chinese Medicine, Zhengzhou 450000, Henan Province, China -
About author:Yu Le, MS, Henan University of Chinese Medicine, Zhengzhou 450000, Henan Province, China -
Supported by:National Natural Science Foundation of China, No. 81573919 (to CYL); Special Research Project on Traditional Chinese Medicine Science and Technology of the National Administration of Traditional Chinese Medicine, No. GZY-KJS-2021-017 (to CYL); Henan Province Key Research and Development Special Project, No. 221111310500 (to CYL); Henan Provincial Science and Technology Key Projects, Nos. 252102311014, 252102310031, and 242102311066 (to CYL)
CLC Number:
Cite this article
Yu Le, Nan Songhua, Shi Zijian, He Qiqi, Li Zhenjia, Cui Yinglin. Mechanisms underlying mitophagy, ferroptosis, cuproptosis, and disulfidptosis in Parkinson’s disease[J]. Chinese Journal of Tissue Engineering Research, 2026, 30(17): 4446-4456.
share this article
Add to citation manager EndNote|Reference Manager|ProCite|BibTeX|RefWorks
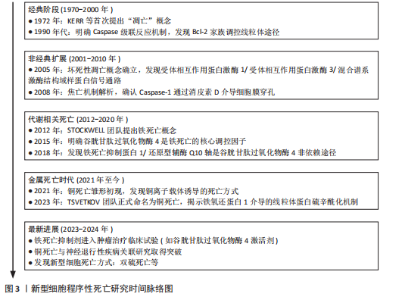
2.1 新型细胞程序性死亡概述 线粒体自噬是通过PTEN诱导激酶1/Parkin通路选择性清除受损线粒体,从而维持能量稳态。在帕金森病中,PTEN诱导激酶1基因突变(如G411S)会导致线粒体自噬受损,进而促进活性氧积累,激活c-Jun氨基末端激酶/p53通路,诱导细胞凋亡[12]。最近的研究发现,Parkin的泛素化修饰能够调控铁死亡关键蛋白谷胱甘肽过氧化物酶4的稳定性,从而形成线粒体自噬与铁死亡之间的交互轴。铁死亡则由System Xc?-谷胱甘肽过氧化物酶4轴的失衡驱动,长链脂酰辅酶A合成酶4/溶血卵磷脂酰基转移酶3通路促进多不饱和脂肪酸的过氧化。在帕金森病中,黑质区的铁过载通过Haber-Weiss反应产生活性氧,并与铜死亡诱导的电子传递链复合体Ⅰ抑制作用相互协作,加剧脂质过氧化[13]。此外核因子E2相关因子2通路的激活可以同时抑制铁死亡和铜死亡,提示抗氧化防御具有全局调控作用。铜死亡依赖于铁氧还蛋白1-二氢硫辛酸转乙酰基酶轴,铜离子直接结合三羧酸循环酶,导致线粒体丙酮酸代谢崩溃和硫辛酰化蛋白聚集。在帕金森病病理进程中已观察到铜离子过度积累现象。铜过载引发Fe-S簇蛋白功能抑制,同时激活长链脂酰辅酶A合成酶4,由此构建了铜-铁代谢的异常循环[14]。线粒体自噬过程的代偿性增强也被证实与此相关。在双硫死亡机制中蛋白质二硫化物异构酶A1-内质网氧化还原酶1信号通路起关键作用。研究显示,细胞骨架蛋白如辅肌动蛋白α4的异常聚集源于二硫键交联反应失调。帕金森病患者存在显著的α-突触核蛋白C端区域中Cys136/Cys145残基的二硫键交联现象,由此引发核因子κB通路激活,随之产生神经炎症反应[15]。值得注意的是,铁死亡标志物谷胱甘肽水平变化能够调控蛋白质二硫化物异构酶A1的氧化还原活性,由此可见双硫死亡与铁死亡间具有潜在联系[16]。作为核心抗氧化枢纽的核因子E2相关因子2通路,其功能具有多重性:谷胱甘肽过氧化物酶4表达上调可抑制铁死亡;铜离子的螯合作用能缓解铜死亡;维持谷胱甘肽水平可以对抗双硫死亡[17-18]。长链脂酰辅酶A合成酶4这一分子靶点值得关注,既参与多不饱和脂肪酸过氧化过程(铁死亡),又能与铜离子结合(铜死亡)。PTEN诱导激酶1/Parkin介导的线粒体自噬功能缺陷时,铁死亡与铜死亡的易感性均会提升。这些证据表明,在帕金森病研究中的4种新型细胞程序性死亡形式(线粒体自噬、铁死亡、铜死亡和双硫死亡)具有重要的研究价值。对这些机制的深入解析不仅能够促进帕金森病病理生理学的认识发展,更为治疗新策略的开发提供可能的分子靶点[19]。 新型程序性死亡时间脉络图见图3。 2.2 线粒体自噬在帕金森病中的作用 2.2.1 线粒体自噬的机制及其功能 研究线粒体自噬机制及其生物学效应对维持线粒体质量具有重要作用[20]。在特定分子标记物及自噬相关蛋白的参与下,功能异常线粒体的选择性清除得以实现[21]。在健康状态下,受损线粒体的标记与包裹过程由PTEN诱导激酶1和Parkin等分子介导完成[22],随后与溶酶体发生融合并降解,该过程不仅消除了异常线粒体,同时参与了细胞能量稳态的调节。氧化应激反应与细胞凋亡调控方面亦显示出重要作用[23]。多项证据表明神经退行性病变、心血管系统疾病乃至恶性肿瘤的发生发展均与线粒体自噬功能障碍存在显著关联[24-25]。以神经退行性疾病为例,阿尔茨海默病与帕金森病等疾病的病理过程中普遍观察到线粒体自噬缺陷。Bcl-2相关卵巢杀手蛋白在Parkin介导的自噬通路中扮演关键角色,研究显示该蛋白缺失会导致线粒体功能障碍及认知能力衰退[26]。在心肌细胞研究中同样发现,通过清除损伤线粒体来维持心脏功能的保护机制依赖于正常的线粒体自噬过程[27-28]。由此可见,深入解析线粒体自噬的分子机制对于疾病治疗策略的开发具有重要价值。后续研究应当着重关注不同疾病背景下线粒体自噬的调控网络及其生物学效应。 2.2.2 线粒体自噬失调与帕金森病的关联 线粒体自噬在帕金森病中的重要性日益受到关注。PTEN诱导激酶1和Parkin是与帕金森病相关的核心蛋白,二者在选择性清除受损线粒体的过程中发挥着重要作用,PTEN诱导激酶1和Parkin突变会导致线粒体自噬功能失调,这与帕金森病的发生紧密相关[29-30]。 线粒体自噬调控失衡已成为帕金森病发病机制的核心环节,PTEN诱导激酶1/Parkin信号轴的功能缺失是导致多巴胺能神经元选择性退变的关键因素。研究显示,赖氨酸乙酰转移酶8介导的Parkin乙酰化修饰对泛素化级联反应具有重要调控作用,其功能缺陷可显著延缓受损线粒体的清除效率[31-32];而赖氨酸乙酰转移酶8调控NSL复合体亚基1通过Unc-51样自噬激活激酶乙酰化调控自噬体形成的分子机制,为解释神经元凋亡特异性增强提供了新视角。近年研究发现,去泛素化酶USP30在帕金森病患者黑质区呈现异常高表达,其通过拮抗Parkin介导的线粒体质量控制,导致线粒体稳态失衡和α-突触核蛋白病理沉积,研究显示:USP30抑制剂(如MTD-101)在动物模型中使线粒体自噬通量提升3倍,但其局限性在于长期抑制可能干扰正常线粒体更新,针对该靶点开发的特异性抑制剂在临床前模型中展现出恢复线粒体自噬通量的治疗潜力,为疾病修饰治疗开辟了新方向[33]。此外,亮氨酸丰富重复激酶2基因突变引发的激酶活性异常,可通过破坏线粒体氧化应激平衡和促进α-突触核蛋白异常聚集双重途径加剧神经退行性变,这也为靶向治疗策略提供了重要分子依据[34-35]。线粒体自噬调控帕金森病的作用机制见图4。 2.2.3 线粒体自噬作为潜在治疗靶点的研究进展 近年研究者们探索了多种药物和治疗策略,以促进线粒体自噬,从而改善帕金森病的病理状态。"
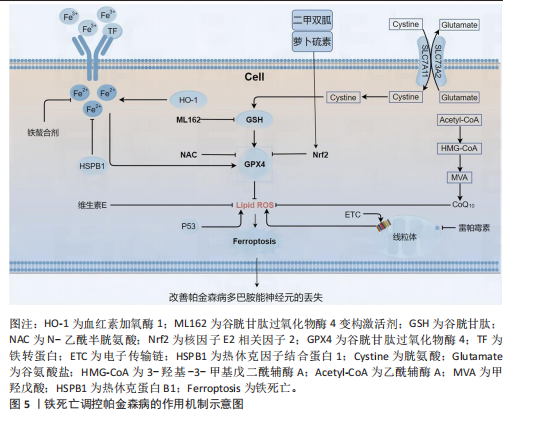
例如伊德本酮(一种合成的辅酶Q10类似物)具有清除自由基、减少脂质过氧化、促进ATP合成等功能,研究显示:在1-甲基-4-苯基-1,2,3,6-四氢吡啶(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,MPTP)诱导的帕金森病小鼠中,伊德本酮使黑质多巴胺能神经元存活率从30%提升至65%,运动功能改善50%。但其局限性在于疗效不足:单药治疗未能逆转疾病进展,可能与晚期病理不可逆相关[36]。在线粒体功能障碍与帕金森病发病机制密切关联的背景下,清除受损线粒体的重要途径——线粒体自噬现象引起了研究者们的广泛关注。作为细胞能量感应器的腺苷单磷酸活化蛋白激酶,在能量不足状态下被激活后,能够促进自噬过程的发生,从而实现对神经细胞的保护效应。具有显著膜穿透特性的蜜蜂毒素的主要成分蜂毒肽,在纳米药物递送系统领域展现出广阔应用前景,研究表明蜂毒肽成功携带siRNA分子穿越血脑屏障。蜂毒肽能够激活腺苷单磷酸活化蛋白激酶/哺乳动物雷帕霉素靶蛋白信号通路,进而促进了线粒体自噬过程。在1-甲基- 4-苯基吡啶离子诱导的SH-SY5Y细胞模型中,蜂毒肽能够改善线粒体稳态。使用Bafilomycin A1自噬抑制剂的结果表明,蜂毒肽的神经保护效应确实依赖于自噬过程的激活[37]。需要引起重视的是蜂毒肽固有的毒性问题亟待通过化学结构修饰予以解决。黄蜂毒液中提取的胡蜂毒液多肽同样表现出了对腺苷单磷酸活化蛋白激酶/哺乳动物雷帕霉素靶蛋白信号通路的激活能力,并促进了线粒体自噬与生物发生过程[38],然而不容忽视的是其潜在的免疫原性问题。这些发现为开发基于天然产物的新型帕金森病治疗策略提供了重要启示。由此可见通过调节腺苷单磷酸活化蛋白激酶和哺乳动物雷帕霉素靶蛋白这两条关键信号通路,线粒体自噬过程可能在帕金森病的治疗中发挥神经保护作用。未来研究应着重探索这一机制的详细分子基础,以期开发出更为有效的治疗新方法。 2.3 铁死亡在帕金森病中的机制 2.3.1 铁死亡的生物学基础 铁死亡是新型程序性细胞死亡形式之一[39],其特征表现为铁依赖性脂质过氧化物累积。区别于凋亡与坏死等其他细胞死亡方式,细胞内铁过度积累及活性氧产生与铁死亡的发生密切相关。多个关键生物过程构成了铁死亡的生物学基础,其中包含铁代谢过程、脂质代谢过程以及抗氧化防御机制系统。在神经退行性疾病特别是帕金森病中发挥着重要作用的是铁死亡,铁的异常堆积是神经元损伤的关键因素之一[40]。谷胱甘肽水平在细胞内的变化、谷胱甘肽过氧化物酶4等抗氧化因子的表达量变化同样对铁死亡的调控过程具有关键影响[41-42]。深入探究铁死亡的分子机制有助于为神经退行性疾病如帕金森病的治疗策略提供潜在的新靶点。 2.3.2 铁代谢异常与帕金森病的关系 帕金森病患者黑质区域中普遍存在铁异常积聚现象。作为神经元损伤的重要诱因,铁的过量堆积不仅导致氧化应激反应的发生,更促进了铁死亡过程的进展性变化。热休克因子结合蛋白1和血红素加氧酶1通过多通路交互作用调控铁死亡与帕金森病病程的发展[43],这一发现具有重要意义。引发脂质过氧化反应的铁积累现象,可观察到多不饱和脂肪酸富集化的特征表现。在神经元膜磷脂中,经由脂氧合酶和花生四烯酸15-脂氧合酶的作用,花生四烯酸与腺苷酰化花生四烯酸-磷脂酰乙醇胺转化为脂质自由基。研究显示脂氧合酶的激活作用会引起α-突触核蛋白聚集化的形成,同时诱导花生四烯酸15-脂氧合酶发生膜转位现象并催化多不饱和脂肪酸的氧化过程。线粒体电子传递链复合物Ⅰ的抑制效应(类似鱼藤酮作用机制)增加了超氧阴离子的生成量[44-45],这一机制可解释细胞膜及细胞器受损的病理过程。转铁蛋白和铁调素等铁代谢相关蛋白的功能失调性表现在帕金森病发病机制中具有显著影响,这些蛋白的表达异常可能加剧铁的积累程度[46],由此可见针对铁代谢异常的干预性措施可能成为帕金森病治疗的新策略方向。 2.3.3 针对铁死亡的干预策略及其研究进展 铁死亡作为一种铁依赖性脂质过氧化驱动的细胞死亡形式,在帕金森病多巴胺能神经元丢失中发挥着关键作用。目前的干预策略主要集中在铁代谢调控与抗氧化防御增强两个方向。 首先,在铁代谢调控策略方面,铁螯合剂去铁胺通过结合游离铁来抑制芬顿反应,临床前研究显示其能够降低帕金森病模型中黑质的铁沉积,并减少α-突触核蛋白寡聚体的形成[47],但长期使用有贫血风险。在铁转运调控方面,抑制转铁蛋白受体1可减少铁的摄入,siRNA沉默铁转蛋白受体1使神经元存活率提升62%。 其次,激活抗氧化通路,谷胱甘肽系统的前体补充,如N-乙酰半胱氨酸,能够绕过胱氨酸/谷氨酸反向转运系统的限制,促进谷胱甘肽合成。脂溶性抗氧化剂如维生素E嵌入细胞膜,阻断脂质过氧化链式反应,延缓帕金森病模型的运动障碍进展[48]。辅酶Q10通过稳定线粒体电子传递链复合体减少活性氧的生成,但其血脑屏障穿透率仅为5%-8%,限制了它在中枢神经系统的疗效。在核因子E2相关因子2通路激活方面,萝卜硫素能够诱导核因子E2相关因子2核转位,并上调血红素加氧酶1、NAD(P)H醌氧化还原酶1等抗氧化基因,临床前研究显示其可减少黑质神经元的丢失。二甲双胍通过腺苷酸活化蛋白激酶/核因子E2相关因子2轴增强转录活性,目前正在进行帕金森病的预防性临床试验。 第三,新型靶向策略中,谷胱甘肽过氧化物酶4变构激活剂如ML162等小分子通过稳定谷胱甘肽过氧化酶4活性中心的构象进而提升酶活性,在MPTP模型中使多巴胺水平得到改善。在铁死亡与自噬的协同调控方面,自噬诱导剂雷帕霉素通过清除受损线粒体间接抑制铁死亡,联合铁螯合剂可使神经元的存活率有效提升。在纳米递送系统方面,脂质体包载的铁抑素1突破血脑屏障,在6-羟基多巴胺模型中使酪氨酸羟化酶阳性细胞数量显著提升[49-50]。 目前面临的挑战与展望:基于脑脊液铁含量(QSM-MRI)和谷胱甘肽过氧化物酶4活性进行分层治疗可能提升疗效;不良反应管控:需要开发组织特异性的铁死亡调节剂,如神经元靶向的小干扰RNA递送系统;临床转化的瓶颈:现有抗氧化剂的中枢生物利用度低(如维生素E < 10%),而纳米载体和前药设计是突破的方向[51-52]。 总之,铁死亡的干预策略已从单一靶点向多通路的协同调控发展,未来需要结合生物标志物的动态监测和精准递送技术,以推动帕金森病修饰治疗的临床转化。铁死亡调控帕金森病作用机制见图5。 2.4 铜死亡在帕金森病中的影响 2.4.1 铜代谢与神经元功能 铜元素作为人体必需微量组分,其生理学意义不容忽视。铜元素参与能量产生过程、神经递质生物合成以及抗氧化系统调控等关键生命活动。当铜代谢稳态遭到破坏时,特别是发生细胞内铜超载现象时,神经细胞将面临毒性威胁。研究显示在帕金森病患者群体中,铜稳态失调与疾病发生发展存在明确关联性[53]。异常升高的铜浓度可能诱发氧化损伤反应、线粒体功能异常乃至最终细胞凋亡等系列病理改变[54]。研究显示与铁代谢网络存在交叉互作的铜代谢途径,其紊乱状态可能协同加剧神经细胞损伤程度[55]。由此可见,保持铜离子浓度处于生理范围对神经细胞存活与功能维持具有决定性影响。适量存在的铜离子不仅保障基础神经活动正常进行,更可能通过特定机制实现对神经组织的保护效应。 2.4.2 铜死亡机制及在帕金森病中的作用 铜稳态失衡现象已被证实与神经退行性病变的发生发展存在关联性。帕金森病患者脑组织中铜含量显著降低,神经元死亡过程及病理学改变与铜含量降低有明显相关性。不仅神经细胞存活状态受到铜代谢紊乱的影响,氧化应激反应的增强以及线粒体功能异常改变也会加速神经退行性病变进程[56]。脂酰化成分在三羧酸循环中的异常结合构成了铜死亡的核心机制,由此引发的细胞内蛋白质毒性应激最终导致细胞死亡的发生。区别于传统凋亡与坏死途径,这种特殊的细胞死亡形式展现出独特的生化标"
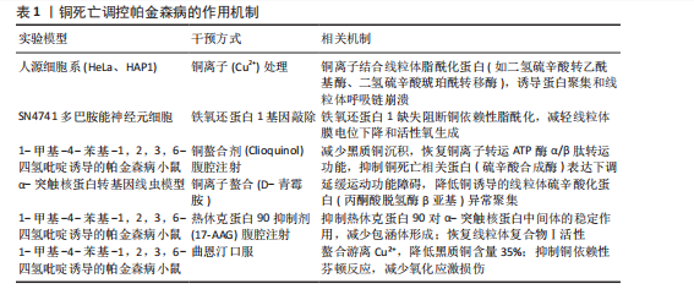
记物特征[57-58]。多巴胺能神经元凋亡过程可能与帕金森病中的铜死亡存在直接联系,该过程受到3个关键因素影响:铜离子缺乏状态、谷胱甘肽含量下降以及线粒体功能受损[59-60]。临床观察与动物实验显示通过干预铜代谢状态可能成为帕金森病治疗新途 径[61-62]。正在进行中的临床试验致力于评估铜调节疗法在阿尔茨海默病及亨廷顿舞蹈症等其他神经退行性疾病中的疗效[63-64]。由此可见铜死亡在帕金森病中的作用机制仍需开展更深入的探究工作,以期获得具有临床应用价值的治疗策略。后续研究重点应当集中于2个方面:铜代谢调控网络的解析及其在神经保护效应中的潜在应用前景。 2.4.3 铜死亡相关的治疗潜力 经Cu2+处理过的人源细胞系(如HeLa与HAP1型),其线粒体脂酰化蛋白组分(包括二氢硫辛酸转乙酰基酶及二氢硫辛酸琥珀酰转移酶等)与铜离子发生特异性结合现象,进而引发蛋白质异常聚集并最终导致线粒体呼吸链功能受损[65]。SN4741型多巴胺能神经元中的铁氧还蛋白1基因被敲除后,铜依赖性脂酰化过程呈现明显阻断态势,同时线粒体膜电位下降幅度及活性氧生成量均有所减轻。在动物模型层面,采用MPTP诱导的帕金森病小鼠经铜螯合剂(例如Clioquinol类)干预后,中脑黑质区的铜沉积量显著降低。与此同时铜离子转运ATP酶α/β肽转运系统的功能性得到恢复,且与铜死亡相关的硫辛酸合成酶蛋白表达水平呈现下调趋势[66]。由此可见此类螯合剂的应用不仅提升了多巴胺能神经元的存活比例,更改善了实验对象的运动功能表现[54]。同时铜螯合剂的实际应用仍存在诸多制约因素,典型表现为中枢神经系统渗透能力较弱——以曲恩汀为例,其在脑组织中的浓度仅维持在血浆浓度的5%-10%范围内,长期给药还可能导致锌、铁等必需微量元素的缺乏症状。铜螯合过程可能干扰超氧化物歧化酶1、环氧化酶等铜依赖性酶的生理活性,从而加重能量代谢紊乱程度。近期研究揭示热休克蛋白家族成员在调控α-突触核蛋白聚集及维持细胞存活方面展现出潜在的治疗价值[67]。基于现有治疗策略的整合优化,通过精确靶向铜代谢相关信号传导网络的新型干预手段或将成为未来帕金森病治疗的重要突破口。这些发现不仅为深入解析帕金森病的发病机制提供了崭新视角,更为创新性治疗方案的开发奠定了坚实的理论基础。铜死亡调控帕金森病的作用机制见表1。 2.5 双硫死亡在帕金森病中的角色 2.5.1 双硫死亡的定义及机制 双硫死亡也是新型细胞死亡模式,其分子机制涉及细胞内双硫键的断裂与形成过程[68]。区别于凋亡、坏死及铁死亡等已知细胞死亡类型,双硫死亡的显著特征在于对硫化物浓度变化和氧化还原稳态的依赖性。在帕金森病病理条件下,神经元的存活状态与功能表现可能受到双硫死亡对氧化还原平衡调节作用的显著影响。线粒体功能障碍现象、氧化应激反应以及铁代谢异常情况与双硫死亡过程具有密切关联性[69],这些病理因素的重要性在帕金森病疾病进展中已被多项研究所证实[59,70]。 2.5.2 双硫死亡与神经退行性病变的关系 在帕金森病病理学进程中,神经元死亡现象及功能异常与双硫死亡机制具有显著相关性。帕金森病患者脑组织中存在氧化应激反应增强和铁元素异常沉积现象,这些病理变化或可成为双硫死亡发生的潜在促进因子。细胞内脂质过氧化作用被双硫死亡所加剧,活性氧簇生成量随之上升,神经细胞损伤程度因而加重。值得注意的是,α-突触核蛋白聚集体的形成与双硫死亡存在关联性,该蛋白异常聚集作为帕金森病典型病理特征而广受关注[71-72]。由此可见深入探究双硫死亡的分子机制学特征,或将开辟帕金森病治疗学研究的新途径。 2.5.3 双硫死亡相关研究的最新进展 近年来,关于双硫死亡现象在帕金森病发生发展过程中的作用机制性研究,逐渐受到学者们的广泛关注。值得注意的是,多项实验性研究已证实,当对双硫死亡相关信号通路进行特异性抑制时,能够显著改善帕金森病模型小鼠的运动功能缺陷和神经元损伤程度。在神经科学研究领域,6-羟基多巴胺和MPTP这两种化学物质被普遍应用于构建帕金森病动物模型,在这些模型中观察到双硫死亡标志物的表达水平变化与神经元损伤程度呈现"
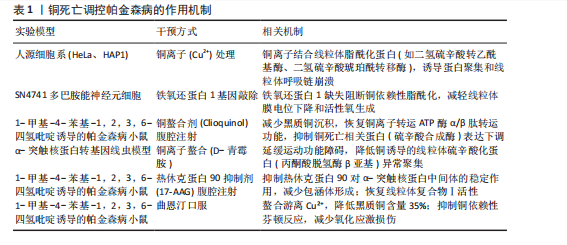
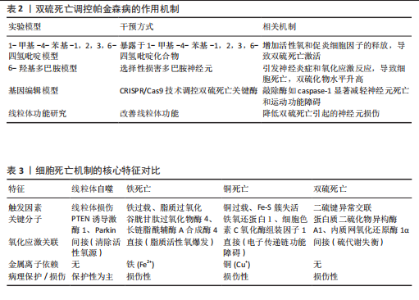
出明显的相关性。在MPTP诱导的帕金森病模型中,不仅表现出严重的运动协调障碍和多巴胺能神经元数量减少,同时伴随着双硫死亡相关蛋白表达量显著上调。MPTP诱导的神经毒性作用通过促进活性氧自由基和促炎性细胞因子的释放,进而激活了双硫死亡通路,加重了神经元的功能异常表现,更推动了帕金森病的疾病进程发展[73]。在6-羟基多巴胺诱导的帕金森病模型中,6-羟基多巴胺通过特异性损伤多巴胺能神经元系统,诱发神经炎症反应和氧化应激状态,细胞内二硫化物含量明显升高,表明双硫死亡途径已被激活。进一步研究发现,核因子κB信号通路和丝裂原活化蛋白激酶级联反应的异常活化在6-羟基多巴胺诱导的细胞死亡过程中发挥着关键性作用。采用基因编辑技术的研究方法为深入理解双硫死亡的分子机制提供了新的可能性。研究显示,通过CRISPR/Cas9系统对caspase-1等关键调控酶进行基因敲除后,实验动物的神经元存活率和运动功能均获得显著改善。这种基因操作手段不仅深化了对双硫死亡分子机制的认识维度,更为开发新型治疗策略开辟了潜在途径。线粒体功能障碍与双硫死亡之间的关联性也在近期研究中得到验证。实验证据表明,通过改善线粒体功能状态可有效减轻由双硫死亡导致的神经元损伤程度。这些研究成果为阐释双硫死亡在帕金森病发病过程中的作用提供了新的理论依据[74-75]。双硫死亡调控帕金森病的作用机制见表2。 2.6 线粒体自噬、铁死亡、铜死亡与双硫死亡的相互关系 2.6.1 各种细胞死亡机制的交互作用与异同点 线粒体自噬、铁死亡、铜死亡与双硫死亡虽然都是细胞死亡机制,但它们的触发条件、分子通路及病理效应存在显著差异(表3)。线粒体自噬的核心功能是通过PTEN诱导激酶1/Parkin通路选择性清除受损线粒体,从而维持能量代谢与氧化还原稳态[76]。线粒体自噬的保护作用是能够抑制其他死亡机制,例如通过清除产生活性氧的线粒体,进而阻断铁死亡。铁死亡的关键驱动因素是铁依赖的脂质过氧化,这一过程由长链脂酰辅酶A合成酶4和溶血卵磷脂酰基转移酶3介导,导致多不饱和脂肪酸的氧化,同时谷胱甘肽过氧化物酶4失活也是其重要特征[77]。铜死亡是一个新机制,铜离子(Cu2+)结合三羧酸循环中的脂酰化蛋白(如铁氧还蛋白1),导致蛋白质的毒性聚集以及Fe-S簇蛋白的失活。铜过载通过抑制Fe-S簇蛋白(例如电子传递链复合体Ⅰ)加剧了线粒体活性氧的爆发,从而形成了与铁死亡之间的“金属毒性循环”[78]。双硫死亡的触发条件是内质网的氧化应激,这导致蛋白二硫键的异常交联,例如α-突触核蛋白的寡聚化,进而阻断自噬溶酶体的降解。它与铁死亡之间存在协同效应,二者共享硫代谢失衡的特征,例如胱氨酸/谷氨酸转运体System Xc?的抑制。 2.6.2 在帕金森病中的协同效应与特殊性 在帕金森病的研究中,正向和负向调控机制对于理解病理特征至关"
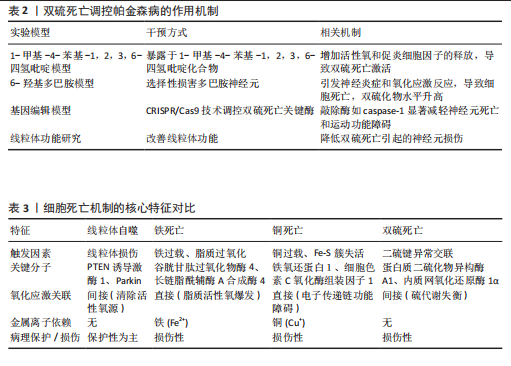

重要。帕金森病的主要病理特征与其他神经退行性疾病存在差异,阿尔茨海默病以β-淀粉样蛋白和tau蛋白缠结为核心,铁死亡与铜死亡主要参与神经元突触丢失,但线粒体自噬缺陷较轻;而亨廷顿病则由突变亨廷顿蛋白介导的线粒体裂变异常主导,铁死亡与双硫死亡的交互较少。帕金森病的特殊性体现于多巴胺能神经元对铁/铜代谢表现出的异常高度敏感性,以及α-突触核蛋白病理学改变与自噬溶酶体系统功能紊乱之间形成的独特恶性循环。选择性丢失在黑质多巴胺能神经元中尤为显著,同时伴随着α-突触核蛋白的异常聚集现象,这些病理特征与铁铜代谢失衡存在着密切关联性。量子相位成像技术(QSM-MRI)结合感应耦合等离子体质谱分析(ICP-MS)的检测数据证实,相较于对照组人群,帕金森病患者黑质区域内铁与铜的含量呈现显著增高趋势[79]。研究显示,当线粒体自噬的正向调控机制出现缺陷时,受损线粒体的积累现象便会发生。大量活性氧物质及游离态铁离子由此释放,脂质过氧化进程被加剧,进而促进铁死亡的发生发展。在PTEN诱导激酶1基因敲除的小鼠模型中观察到黑质多巴胺能神经元的铁死亡水平明显升高。功能正常的线粒体自噬过程能够有效清除那些铁超载的线粒体,降低与铁死亡相关酶类(如长链脂酰辅酶A合成酶4)的激活程度,铁依赖性细胞死亡由此得到抑制。铜代谢异常在负向调控方面展现出特殊作用。电子传递链复合体等铁硫簇蛋白受到抑制后,形成ATP耗竭状态,激活长链脂酰辅酶A合成酶4/溶血卵磷脂酰基转移酶3通路,脂质过氧化物的生成量增加。此外,铁的过量蓄积经由芬顿反应促使羟基自由基(·OH)大量产生,使得铜介导的氧化应激现象更趋恶化,最终形成“金属毒性的正反馈循环”。在帕金森病等多种神经退变疾病中,这一调控性机制可能具有不可忽视的病理学意义。铜元素的这种调控特性在代谢异常疾病与神经退行性病变中显示出潜在重要性。后续研究工作中亟待阐明的是,铜离子与铁硫簇蛋白间的交互作用模式,以及这种相互作用如何在不同生理状态和疾病条件下对细胞功能学特征和代谢通路网络产生影响。上述研究将为揭示铜元素在细胞代谢过程中所扮演的双重性角色提供关键线索[80],并为相关疾病的临床干预策略开辟新途径。由c-Jun氨基末端激酶相互作用蛋白4-瞬时受体电位黏蛋白1-凋亡关联基因2构成的功能性复合体,对溶酶体的空间定位与生物学功能的调控作用能够显著影响自噬过程与铁死亡途径之间的交叉对话。在双硫死亡与铁死亡的相互作用中,蛋白质二硫化物异构酶A1-谷胱甘肽代谢途径展现出特殊重要性[81]。对于细胞内蛋白质二硫键的稳态维持,蛋白质二硫化物异构酶A1发挥着不可替代的作用。还原型谷胱甘肽代谢过程的调控同样受到二硫化物异构酶A1蛋白的影响,细胞抗氧化防御体系因此发生改变。铁死亡的发生发展涉及两个核心要素:铁离子的异常蓄积与活性氧自由基的过量产生。谷胱甘肽水平显著下降导致氧化应激敏感性增加,铁死亡进程加速推进。研究表明,蛋白质二硫化物异构酶A1-谷胱甘肽代谢网络可能构成双硫死亡与铁死亡的交叉调控节点[15]。该代谢途径的分子机制尚待阐明,在各类疾病中的潜在应用价值更需要后续研究予以确认。 2.6.3 靶向协同网络的干预策略 近年来的研究趋势表明,帕金森病的治疗学方法正经历着向多靶向协同干预模式的转型。在临床前阶段的实验观察中,采用的是铁离子螯合剂去铁酮与铜离子螯合剂曲恩汀的联合给药方案,该方案能够使得MPTP诱导的小鼠模型内黑质区神经元的存活数量显著提升[82],这一现象为早期阶段帕金森病患者的临床处理开辟了新颖途径。正在进行中的临床试验针对此种联合用药方案对于早期帕金森病患者黑质区铁沉积状况以及统一帕金森病量表评分的影响程度展开了系统评估[83],其目的在于对临床疗效进行更深入验证。关于自噬过程的激活与抗氧化效应的协同治疗策略,采用雷帕霉素(自噬过程激活物质)联合Ferrostatin-1 (铁死亡抑制化合物)的给药方式,在6-羟基多巴胺动物模型中观察到了明显的治疗效果[84],体现在脂质过氧化产物水平出现下降趋势,同时多巴胺类神经递质水平得以恢复。值得关注的是,针对长链脂酰辅酶A合成酶4-溶血卵磷脂酰基转移酶3信号转导通路的小分子抑制物质(例如PRGL493类化合物),在铜离子过载的细胞实验体系中也展示出良好的干预效果[85],其能够有效抑制脂质过氧化反应进程,并减少α-突触核蛋白寡聚体的生成数量。随着对帕金森病病理学机制认识的不断深化,越来越多的科研工作开始聚焦于传统中医药理论的干预策略。部分中药活性成分(如黄酮类化合物、生物碱类物质以及多糖类成分等)被证实具有明显的神经保护特性[86-87],这些物质通过抗氧化应激、抑制炎症反应等多重作用机制实现神经功能的改善目标。综合现有研究证据可以认为,帕金森病的治疗策略正在朝着多靶向干预和个体化医疗的方向演进。将现代医学技术与传统中医理论的优势特点进行有机结合,预期将为临床患者带来更为有效的治疗方案选择。"

| [1] TIAN HY, HUANG BY, NIE HF, et al. The Interplay between Mitochondrial Dysfunction and Ferroptosis during Ischemia-Associated Central Nervous System Diseases. Brain Sci. 2023;13(10):1367. [2] 范平龙,赖华清,张钊,等.缺血性脑卒中后小胶质细胞胞葬作用的研究进展[J].中国药理学通报,2024,40(8):1407-1412. [3] WANG XX, LI M, XU XW, et al. BNIP3-mediated mitophagy attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis through P62-KEAP1-NRF2 pathway activation to maintain iron and redox homeostasis. Acta Pharmacol Sin. 2025;46(1):33-51. [4] 郭雪微. PIEZO1调控急性损伤致肺内皮细胞铁死亡的机制研究[D].锦州:锦州医科大学,2022. [5] LI J, JIA YC, DING YX, et al. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Biol Sci. 2023;19(9):2756-2771. [6] WANG Y, YAN Q, SHI Y, et al. Copper Toxicity in Animals: A Review. Biol Trace Elem Res. 2025;203(5):2675-2686. [7] 邹鑫.铁死亡相关基因FDFT1可作为肾透明细胞癌的潜在生物标志物[D].南昌:南昌大学,2023. [8] 易威威. HO-1通过诱导线粒体途径的自噬来抑制人髓核细胞衰老和退变[D]. 重庆:重庆医科大学,2020. [9] LIU Y, LU S, WU LL, et al. The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. 2023;14(8):519. [10] WANG W, LU K, JIANG X, et al. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J Exp Clin Cancer Res. 2023;42(1):142. [11] 刘娟,李彦杰,秦合伟,等.线粒体质量控制系统失调介导帕金森病的作用机制[J].实用医学杂志,2024,40(11):1479-1482. [12] KREIMENDAHL S, RASSOW J. The Mitochondrial Outer Membrane Protein Tom70-Mediator in Protein Traffic, Membrane Contact Sites and Innate Immunity. Int J Mol Sci. 2020;21(19):7262. [13] FERREIRA C, VIANA SD, REIS F. Gut Microbiota Dysbiosis-Immune Hyperresponse-Inflammation Triad in Coronavirus Disease 2019 (COVID-19): Impact of Pharmacological and Nutraceutical Approaches. Microorganisms. 2020;8(10):1514. [14] LIANG Y, MENG Z, CHEN Y, et al. A Data Fusion Orientation Algorithm Based on the Weighted Histogram Statistics for Vector Hydrophone Vertical Array. Sensors (Basel). 2020;20(19):5619. [15] ZHU YY, ZHANG Q, JIA YC, et al. Protein disulfide isomerase plays a crucial role in mediating chemically-induced, glutathione depletion-associated hepatocyte injury in vitro and in vivo. Cell Commun Signal. 2024;22(1):431. [16] SCHERGER M, BOLLI E, ANTUNES ARP, et al. Transient Multivalent Nanobody Targeting to CD206-Expressing Cells via PH-Degradable Nanogels. Cells. 2020;9(10):2222. [17] PENG J, DAI X, ZHANG T, et al. Copper as the driver of the lncRNA-TCONS-6251/miR-novel-100/TC2N axis: Unraveling ferroptosis in duck kidney. Int J Biol Macromol. 2024; 282(Pt 2):136797. [18] WANG M, MA F, ZHONG G, et al. Copper exposure promotes ferroptosis of chicken (Gallus gallus) kidney cells and causes kidney injury. J Trace Elem Med Biol. 2024;86:127501. [19] 王晓蓓,麦迪乃·赛福丁,白睿,等.基于孟德尔随机研究血清炎症因子、生化指标及经颅黑质超声在帕金森病患者中的价值[J].齐齐哈尔医学院学报,2024, 45(7):608-613. [20] 于守水.线粒体钙单向转运体对神经母细胞瘤细胞自噬和线粒体自噬的作用[D].青岛:青岛大学,2017. [21] 韦云耿.矽尘诱导肺泡巨噬细胞焦亡与线粒体自噬相互作用调控矽肺纤维化进展的作用与机制研究[D].沈阳:中国医科大学,2023. [22] ONISHI M, YAMANO K, SATO M, et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021; 40(3):e104705. [23] 张莹莹,叶周恒,刘昕,等.SIRT3在心脏衰老中的作用机制[J].生命的化学,2023, 43(2):215-220. [24] PICCA A, FAITG J, AUWERX J, et al. Mitophagy in human health, ageing and disease. Nat Metab. 2023;5(12):2047-2061. [25] DENISENKO TV, GOGVADZE V, ZHIVOTOVSKY B. Mitophagy in carcinogenesis and cancer treatment. Discov Oncol. 2021;12(1):58. [26] YANG Y, CHEN H, HUANG S, et al. BOK-engaged mitophagy alleviates neuropathology in Alzheimer’s disease. Brain. 2025;148(2):432-447. [27] TITUS AS, SUNG EA, ZABLOCKI D, et al. Mitophagy for cardioprotection. Basic Res Cardiol. 2023;118(1):42. [28] SAITO T, HAMANO K, SADOSHIMA J. Molecular mechanisms and clinical implications of multiple forms of mitophagy in the heart. Cardiovasc Res. 2021;117(14): 2730-2741. [29] MASALDAN S, CALLEGARI S, DEWSON G. Therapeutic targeting of mitophagy in Parkinson’s disease. Biochem Soc Trans. 2022;50(2):783-797. [30] SOUTAR MPM, MELANDRI D, O’CALLAGHAN B, et al. Regulation of mitophagy by the NSL complex underlies genetic risk for Parkinson’s disease at 16q11.2 and MAPT H1 loci. Brain. 2022;145(12):4349-4367. [31] PEKER N, DONIPADI V, SHARMA M, et al. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am J Physiol Cell Physiol. 2018;315(2):C164-C185. [32] BLAGOV AV, GONCHAROV AG, BABICH OO, et al. Prospects for the Development of Pink1 and Parkin Activators for the Treatment of Parkinson’s Disease. Pharmaceutics. 2022;14(11):2514. [33] THOBOIS S. USP30: a new promising target for Parkinson’s disease? Mov Disord. 2015; 30(3):340. [34] SINGH F, PRESCOTT AR, ROSEWELL P, et al. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife. 2021;10: e67604. [35] FRANCO F, BEVILACQUA A, WU RM, et al. Regulatory circuits of mitophagy restrict distinct modes of cell death during memory CD8+ T cell formation. Sci Immunol. 2023; 8(87):eadf7579. [36] YAN J, SUN W, SHEN M, et al. Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice. Cell Death Discov. 2022;8(1):28. [37] CHEN M, WANG X, BAO S, et al. Orchestrating AMPK/mTOR signaling to initiate melittin-induced mitophagy: A neuroprotective strategy against Parkinson’s disease. Int J Biol Macromol. 2024;281(Pt 1): 136119. [38] LIU C, LI X, CHEN M, et al. Characterization and neurotherapeutic evaluation of venom polypeptides identified from Vespa magnifica: The role of Mastoparan-M in Parkinson’s disease intervention. J Ethnopharmacol. 2025;343:119481. [39] 李佳蔚,许红阳.铁死亡在器官移植缺血-再灌注损伤中的作用与展望[J].器官移植,2023,14(5):662-668. [40] CHENG X, ZHAO F, KE B, et al. Harnessing Ferroptosis to Overcome Drug Resistance in Colorectal Cancer: Promising Therapeutic Approaches. Cancers (Basel). 2023;15(21):5209. [41] 农复香,蒋志雄,李英豪,等.外泌体调控铁死亡在疾病诊断治疗中的应用与作用[J].中国组织工程研究,2023,27(15): 2443-2452. [42] GAO G, YOU L, ZHANG J, et al. Brain Iron Metabolism, Redox Balance and Neurological Diseases. Antioxidants (Basel). 2023;12(6):1289. [43] YAO Z, JIAO Q, DU X, et al. Ferroptosis in Parkinson’s disease -- The iron-related degenerative disease. Ageing Res Rev. 2024;101:102477. [44] 戴美华.艾司氯胺酮预处理对大鼠脑缺血再灌注损伤的影响及与Nrf2/HO-1信号通路的关系[D].广州:广州医科大学,2023. [45] LIU T, WANG P, YIN H, et al. Rapamycin reverses ferroptosis by increasing autophagy in MPTP/MPP+-induced models of Parkinson’s disease. Neural Regen Res. 2023;18(11):2514-2519. [46] AN F, ZHANG J, GAO P, et al. New insight of the pathogenesis in osteoarthritis: the intricate interplay of ferroptosis and autophagy mediated by mitophagy/chaperone-mediated autophagy. Front Cell Dev Biol. 2023;11:1297024. [47] 孟繁星.颅内铁离子超载在脊髓损伤后中枢性疼痛中的作用研究[D].重庆:第三军医大学,2017. [48] LI T, SUN M, SUN Q, et al. PM2.5-induced iron homeostasis imbalance triggers cardiac hypertrophy through ferroptosis in a selective autophagy crosstalk manner. Redox Biol. 2024;72:103158. [49] PENG Z, DING YN, YANG ZM, et al. Neuron-targeted liposomal coenzyme Q10 attenuates neuronal ferroptosis after subarachnoid hemorrhage by activating the ferroptosis suppressor protein 1/coenzyme Q10 system. Acta Biomater. 2024;179: 325-339. [50] ZHU J, CAI Y, KONG M, et al. Design, Synthesis, and Biological Evaluation for First GPX4 and CDK Dual Inhibitors. J Med Chem. 2024;67(4):2758-2776. [51] LI X, RAN Q, HE X, et al. HO-1 upregulation promotes mitophagy-dependent ferroptosis in PM2.5-exposed hippocampal neurons. Ecotoxicol Environ Saf. 2024;277:116314. [52] LONG Z, LUO Y, YU M, et al. Targeting ferroptosis: a new therapeutic opportunity for kidney diseases. Front Immunol. 2024; 15:1435139. [53] PRADHAN SH, LIU JY, SAYES CM. Evaluating Manganese, Zinc, and Copper Metal Toxicity on SH-SY5Y Cells in Establishing an Idiopathic Parkinson’s Disease Model. Int J Mol Sci. 2023;24(22):16129. [54] SCOLARI GROTTO F, GLASER V. Are high copper levels related to Alzheimer’s and Parkinson’s diseases? A systematic review and meta-analysis of articles published between 2011 and 2022. Biometals. 2024; 37(1):3-22. [55] GROMADZKA G, WILKANIEC A, TARNACKA B, et al. The Role of Glia in Wilson’s Disease: Clinical, Neuroimaging, Neuropathological and Molecular Perspectives. Int J Mol Sci. 2024;25(14):7545. [56] DAVIES KM, MERCER JF, CHEN N, et al. Copper dyshomoeostasis in Parkinson’s disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci (Lond). 2016;130(8):565-574. [57] LIDDELL JR, WHITE AR. Nexus between mitochondrial function, iron, copper and glutathione in Parkinson’s disease. Neurochem Int. 2018;117:126-138. [58] GROMADZKA G, TARNACKA B, FLAGA A, et al. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int J Mol Sci. 2020;21(23):9259. [59] HUANG M, ZHANG Y, LIU X. The mechanism of cuproptosis in Parkinson’s disease. Ageing Res Rev. 2024;95:102214. [60] GIL-BEA FJ, ALDANONDO G, LASA-FERNÁNDEZ H, et al. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert Rev Mol Med. 2017;19:e7. [61] RECZEK CR, BIRSOY K, KONG H, et al. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat Chem Biol. 2017;13(12):1274-1279. [62] FU Y, ZENG S, WANG Z, et al. Mechanisms of Copper-Induced Autophagy and Links with Human Diseases. Pharmaceuticals (Basel). 2025;18(1):99. [63] SCHOLEFIELD M, UNWIN RD, COOPER GJS. Shared perturbations in the metallome and metabolome of Alzheimer’s, Parkinson’s, Huntington’s, and dementia with Lewy bodies: A systematic review. Ageing Res Rev. 2020;63:101152. [64] LAUTERBACH EC. Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification. Neural Regen Res. 2016;11(11):1712-1726. [65] TSVETKOV P, COY S, PETROVA B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254-1261. [66] SONG X, LIU T, YU L, et al. OTUD5 Protects Dopaminergic Neurons by Promoting the Degradation of α-Synuclein in Parkinson’s Disease Model. Adv Sci (Weinh). 2025;12(7):e2406700. [67] LI X, WANG W, PAN S, et al. Exploring heat shock proteins as therapeutic targets for Parkinson’s disease. Biochem Pharmacol. 2024;230(Pt 3):116633. [68] 孙双一,贺新新,陈雯桐,等.双硫死亡相关基因对结直肠癌预后和药物敏感性的预测价值[J].复旦学报(医学版), 2024.51(4):473-483. [69] 李梦.皮肤活检评价自噬异常调控参与帕金森病发病机制的研究[D].郑州:郑州大学,2014. [70] DING XS, GAO L, HAN Z, et al. Ferroptosis in Parkinson’s disease: Molecular mechanisms and therapeutic potential. Ageing Res Rev. 2023;91:102077. [71] YANG K, ZENG L, ZENG J, et al. Research progress in the molecular mechanism of ferroptosis in Parkinson’s disease and regulation by natural plant products. Ageing Res Rev. 2023;91:102063. [72] TIAN Y, LU J, HAO X, et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics. 2020;17(4):1796-1812. [73] WANG G, ZHUANG W, ZHOU Y, et al. 17β-estradiol alleviated ferroptotic neuroinflammation by suppressing ATF4 in mouse model of Parkinson’s disease. Cell Death Discov. 2024;10(1):507. [74] LIU C, LIU Z, FANG Y, et al. Exposure to the environmentally toxic pesticide maneb induces Parkinson’s disease-like neurotoxicity in mice: A combined proteomic and metabolomic analysis. Chemosphere. 2022;308(Pt 2):136344. [75] ZHOU M, XU K, GE J, et al. Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies. Int J Mol Sci. 2024;25(23):13042. [76] 何云凌.低氧下BNIP3翻译后修饰对线粒体自噬的调控作用[D].北京:军事科学院,2018. [77] 裴卓.铁死亡调控特发性肺纤维化发生发展的分子机制研究[D].西安:中国人民解放军空军军医大学,2022. [78] 张颖.铜暴露诱导认知障碍及神经毒性的机制研究[D].南京:东南大学,2022. [79] LI Y, SUN W, YUAN S, et al. The role of cuproptosis in gastric cancer. Front Immunol. 2024;15:1435651. [80] BELLOLI S, MORARI M, MURTAJ V, et al. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front Aging Neurosci. 2020;12:152. [81] TANG YH, WU L, HUANG HL, et al. Hydrogen sulfide antagonizes formaldehyde-induced ferroptosis via preventing ferritinophagy by upregulation of GDF11 in HT22 cells. Toxicology. 2023; 491:153517. [82] FOLTYNIE T, GANDHI S, GONZALEZ-ROBLES C, et al. Towards a multi-arm multi-stage platform trial of disease modifying approaches in Parkinson’s disease. Brain. 2023;146(7):2717-2722. [83] JING H, WANG S, WANG M, et al. Isobavachalcone Attenuates MPTP-Induced Parkinson’s Disease in Mice by Inhibition of Microglial Activation through NF-κB Pathway. PLoS One. 2017; 12(1):e0169560. [84] LIU X, LIU Y, LIU J, et al. Correlation between the gut microbiome and neurodegenerative diseases: a review of metagenomics evidence. Neural Regen Res. 2024;19(4):833-845. [85] OUTEIRO TF, HARVEY K, DOMINGUEZ-MEIJIDE A, et al. LRRK2, alpha-synuclein, and tau: partners in crime or unfortunate bystanders? Biochem Soc Trans. 2019; 47(3):827-838. [86] MENG W, CHAO W, KAIWEI Z, et al. Bioactive compounds from Chinese herbal plants for neurological health: mechanisms, pathways, and functional food applications. Front Nutr. 2025;12:1537363. [87] KUJAWSKA M, JODYNIS-LIEBERT J. Polyphenols in Parkinson’s Disease: A Systematic Review of In Vivo Studies. Nutrients. 2018;10(5):642. |
| No related articles found! |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||