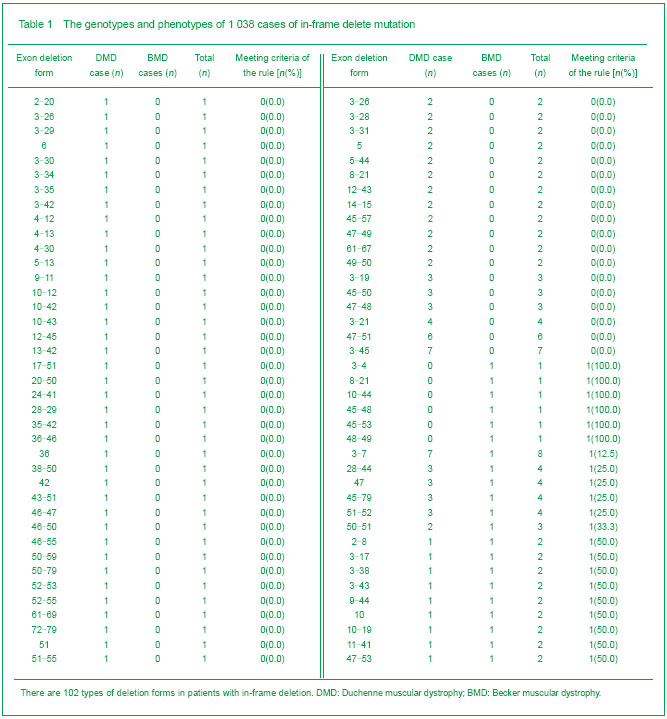
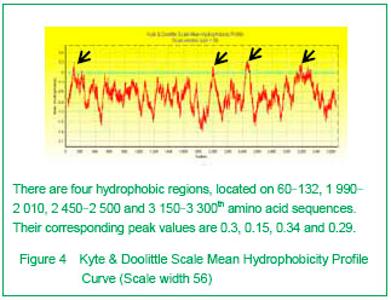
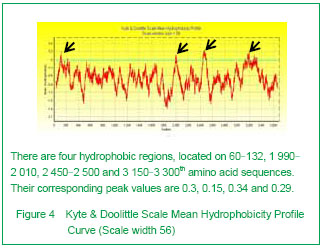
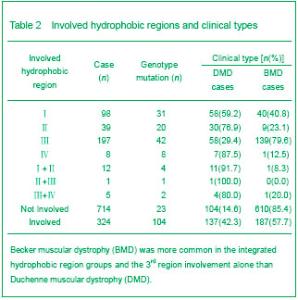
| [1] Pane M, Lombardo ME, Alfieri P, et al. Attention deficit hyperactivity disorder and cognitive function in Duchenne muscular dystrophy: phenotype-genotype correlation. J Pediatr. 2012;161(4):705-709.e1.[2] Magri F, Govoni A, D'Angelo MG, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258(9): 1610-1623.[3] Escolar DM, Zimmerman A, Bertorini T, et al. Pentoxifylline as a rescue treatment for DMD: a randomized double-blind clinical trial. Neurology. 2012;78(12):904-913. [4] López-Hernández LB, van Heusden D, Soriano-Ursúa MA, et al. Genotype-phenotype discordance in a Duchenne muscular dystrophy patient due to a novel mutation: insights into the shock absorber function of dystrophin. Rev Neurol. 2011;52(12):720-724.[5] Bushby K, Connor E. Clinical outcome measures for trials in Duchenne muscular dystrophy: report from International Working Group meetings. Clin Investig (Lond). 2011;1(9): 1217-1235.[6] Scully MA, Cwik VA, Marshall BC, et al. Can outcomes in Duchenne muscular dystrophy be improved by public reporting of data? Neurology. 2013;80(6):583-589. [7] Soltanzadeh P, Friez MJ, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord. 2010;20(8):499-504. [8] Basumatary LJ, Das M, Goswami M, et al. Deletion pattern in the dystrophin gene in Duchenne muscular dystrophy patients in northeast India. J Neurosci Rural Pract. 2013; 4(2):227-229. [9] Moore CJ, Winder SJ. The inside and out of dystroglycan post-translational modification. Neuromuscul Disord. 2012; 22(11):959-965. [10] Janke A, Upadhaya R, Snow WM, et al. A new look at cytoskeletal NOS-1 and β-dystroglycan changes in developing muscle and brain in control and mdx dystrophic mice. Dev Dyn. 2013. in press.[11] Kalman L, Leonard J, Gerry N, et al. Quality assurance for Duchenne and Becker muscular dystrophy genetic testing: development of a genomic DNA reference material panel. J Mol Diagn. 2011;13(2):167-174. [12] Rani AQ, Sasongko TH, Sulong S, et al. Mutation spectrum of dystrophin gene in malaysian patients with Duchenne/Becker muscular dystrophy. J Neurogenet. 2013;27(1-2):11-15.[13] Lee BL, Nam SH, Lee JH, et al. Genetic analysis of dystrophin gene for affected male and female carriers with Duchenne/Becker muscular dystrophy in Korea. J Korean Med Sci. 2012;27(3):274-280. [14] de Brouwer AP, Nabuurs SB, Verhaart IE, et al. A 3-base pair deletion, c.9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur J Hum Genet. 2013. in press.[15] Kesari A, Pirra LN, Bremadesam L, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum Mutat. 2008;29(5):728-737. [16] Zhang D, Cui S, Guo H, et al. Genomic structure, characterization and expression analysis of a manganese superoxide dismutase from pearl oyster Pinctada fucata. Dev Comp Immunol. 2013;41(4):484-490. [17] Yang J, Li SY, Li YQ, et al. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet. 2013;14:29. [18] Tuffery-Giraud S, Béroud C, Leturcq F, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6): 934-945. [19] Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1): 105-132.[20] Ghose S, Tao Y, Conley L, et al. Purification of monoclonal antibodies by hydrophobic interaction chromatography under no-salt conditions. MAbs. 2013;5(5):795-800. [21] Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an "LSDB-in-a-box" approach. Hum Mutat. 2005;26(2):63-68.[22] Fokkema IF, Taschner PE, Schaafsma GC, et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557-563. [23] Bushby KM, Gardner-Medwin D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. 1993;240(2):98-104.[24] Norwood FL, Sutherland-Smith AJ, Keep NH, et al. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000; 8(5):481-491.[25] Zhang C, Cai JH, Liu ZL, et al. The relationship between dystrophin hydrophobic structure and DMD/BMD. Zhongshan Yike Daxue Xuebao. 1993;14(2):160. [26] Kiefer F, Arnold K, Künzli M, et al. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37(Database issue):D387-392. [27] Goodsell DS. Representing structural information with RasMol. Curr Protoc Bioinformatics. 2005;Chapter 5:Unit 5.4.[28] Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein. II. Structure determination and general description. J Biol Chem. 1973;248(9):3313-3326.[29] Heidarsson PO, Otazo MR, Bellucci L, et al. Single-Molecule Folding Mechanism of an EF-Hand Neuronal Calcium Sensor. Structure. 2013;21(10):1812-1821. [30] Bork P, Sudol M. The WW domain: a signalling site in dystrophin? Trends Biochem Sci. 1994;19(12):531-533.[31] Hiraki T, Abe F. Overexpression of Sna3 stabilizes tryptophan permease Tat2, potentially competing for the WW domain of Rsp5 ubiquitin ligase with its binding protein Bul1. FEBS Lett. 2010;584(1):55-60. [32] Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53(2):219-228.[33] Gumerson JD, Michele DE. The dystrophin-glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;2011:210797.[34] Mondal J, Morrone JA, Berne BJ. How hydrophobic drying forces impact the kinetics of molecular recognition. Proc Natl Acad Sci U S A. 2013;110(33):13277-13282.[35] Norwood FL, Sutherland-Smith AJ, Keep NH, et al. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure. 2000; 8(5): 481-491.[36] Singh SM, Mallela KM. The N-terminal actin-binding tandem calponin-homology (CH) domain of dystrophin is in a closed conformation in solution and when bound to F-actin. Biophys J. 2012;103(9):1970-1978. [37] Legrand B, Giudice E, Nicolas A, et al. Computational study of the human dystrophin repeats: interaction properties and molecular dynamics. PLoS One. 2011;6(8):e23819. [38] Acsadi G, Moore SA, Chéron A, et al. Novel mutation in spectrin-like repeat 1 of dystrophin central domain causes protein misfolding and mild Becker muscular dystrophy. J Biol Chem. 2012;287(22):18153-18162.[39] Mirza A, Menhart N. Stability of dystrophin STR fragments in relation to junction helicity. Biochim Biophys Acta. 2008; 1784(9):1301-1309.[40] Lai Y, Zhao J, Yue Y, et al. α2 and α3 helices of dystrophin R16 and R17 frame a microdomain in the α1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A. 2013;110(2):525-530. [41] Harper SQ. Molecular dissection of dystrophin identifies the docking site for nNOS. Proc Natl Acad Sci U S A. 2013; 110(2):387-388. [42] Cazzella V, Martone J, Pinnarò C, et al. Exon 45 skipping through U1-snRNA antisense molecules recovers the Dys-nNOS pathway and muscle differentiation in human DMD myoblasts. Mol Ther. 2012;20(11):2134-2142. [43] Sahni N, Mangat K, Le Rumeur E, et al. Exon edited dystrophin rods in the hinge 3 region. Biochim Biophys Acta. 2012;1824(10):1080-1089. [44] Banks GB, Judge LM, Allen JM, et al. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6(5):e1000958.[45] Carsana A, Frisso G, Tremolaterra MR, et al. Analysis of dystrophin gene deletions indicates that the hinge III region of the protein correlates with disease severity. Ann Hum Genet. 2005;69(Pt 3):253-259. [46] Nicholson LV, Johnson MA, Bushby KM, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 2. Correlations within individual patients. J Med Genet. 1993;30(9):737-744.[47] Stahelin RV. Lipid binding domains: more than simple lipid effectors. J Lipid Res. 2009;50 Suppl:S299-304. [48] Aoki Y, Nakamura A, Yokota T, et al. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol Ther. 2010;18(11):1995-2005. [49] Chung W, Campanelli JT. WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol Cell Biol Res Commun. 1999;2(3):162-171.[50] Hnia K, Zouiten D, Cantel S, et al. ZZ domain of dystrophin and utrophin: topology and mapping of a beta-dystroglycan interaction site. Biochem J. 2007;401(3):667-677. [51] Huang X, Poy F, Zhang R, et al. Structure of a WW domain containing fragment of dystrophin in complex with beta-dystroglycan. Nat Struct Biol. 2000;7(8):634-638.[52] Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta. 2013. in press.[53] Cirak S, Feng L, Anthony K, et al. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20(2):462-467. [54] R Mendell J, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013. in press. |