Chinese Journal of Tissue Engineering Research ›› 2019, Vol. 23 ›› Issue (24): 3882-3888.doi: 10.3969/j.issn.2095-4344.1298
Previous Articles Next Articles
Molecular biological characteristics of hereditary multiple exostoses
- Department of Orthopedics, Changhai Hospital, Navy Medical University, Shanghai 200433, China
-
Online:2019-08-28Published:2019-08-28 -
Contact:He Dawei, MD, Chief physician, Professor, Department of Orthopedics, Changhai Hospital, Navy Medical University, Shanghai 200433, China -
About author:Wu Donghua, Master, Physician, Department of Orthopedics, Changhai Hospital, Navy Medical University, Shanghai 200433, China -
Supported by:the National Natural Science Foundation of China, No. 8157100974 (to HDW)
CLC Number:
Cite this article
Wu Donghua, He Dawei .
share this article
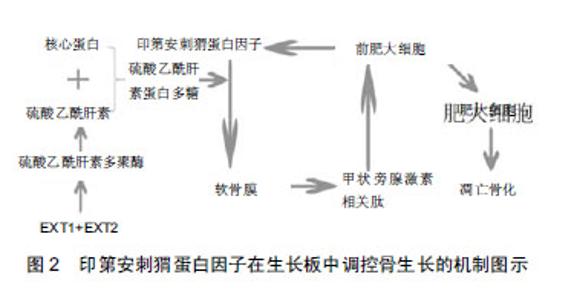
2.1 概述 骨软骨瘤是最常见的良性骨肿瘤,占骨肿瘤总数的18.4%,多为单发,多发者通常有家族史,称之为遗传性多发性骨软骨瘤。世界卫生组织定义其为宽基底(无蒂)或者茎状(有蒂)与肢体骨髓腔相通,表面覆盖软骨帽,由骨皮质构成的外生骨性凸起。1814年Bover首先报道了多个遗传性多发性骨软骨瘤患者的一个家系。1964年Solomom[1]最先从基因角度研究该疾病。 2.2 病理改变 骨软骨瘤的特征性表现为在长骨的干骺端与骨干移行部,有软骨帽的骨块凸起于关节,其下骨皮质和骨软骨瘤茎的皮质相延续,其内填充松质骨。在光镜下可见特征性的3层结构,从上到下依次为软骨膜、软骨帽和骨[2]。软骨层可见呈柱状排列的软骨细胞,软骨/骨交界处发生软骨内骨化,可见双核细胞、钙化、坏死、结节和囊性改变等。瘤体持续生长依赖于软骨帽的软骨化骨作用[3],软骨帽厚度的增厚缘由软骨帽瘤细胞体积的增大及细胞数量的增加[4]。瘤细胞由单突变细胞克隆增殖而来[5]。观察软骨帽的超微结构可见软骨组织内有大量瘤样细胞增生,呈聚集分布,细胞核较大,细胞质内可见圆形或椭圆形的线粒体及扩张的粗面内质网;瘤细胞间可见毛细血管,附近的软骨细胞明显增多,软骨细胞体积较正常细胞增大[6]。在成人,软骨帽的厚度通常小于1 cm,当软骨帽超过1.5-2.0 cm或肿块所在部位持续性疼痛及肿块停止生长后再次增大,则提示肿瘤发生恶变,此时软骨细胞增多、胞体肥大、核大深染,可见双核瘤细胞和多核瘤巨细胞及病理性核分裂象[7]。遗传性多发性骨软骨瘤通常恶变为软骨肉瘤,发生率为0.5%-2.0%,少见恶变为骨肉瘤和纤维肉瘤[8-9]。相对于原发恶性肿瘤,遗传性多发性骨软骨瘤患者恶变多为低级别软骨肉瘤,生长缓慢,很少发生转移,预后良好。大约10%的骨软骨瘤恶变为去分化软骨肉瘤及非软骨高级别肉瘤2类,均具有较高的侵蚀性,容易转移且预后较差[10]。 2.3 与骨软骨瘤发生相关的骨生长发育因素 骨和软骨组织属于结缔组织的特殊类型,最初由胚胎时期外胚层和内胚层之间的间充质分化而来。骨和软骨均有膜下及软骨内2种生长方式,遗传性多发性骨软骨瘤发病主要见于软骨内成骨的骨结构,在将要成骨的部位,间充质聚集,分化为骨祖细胞,再分化为软骨细胞,软骨细胞分泌软骨基质,将细胞包埋,形成软骨组织,周围的间充质分化为软骨膜,形成一块软骨,中段软骨膜内的骨祖细胞分化为成骨细胞,包绕软骨中段,形成骨领,中央的软骨基质钙化、软骨细胞凋亡,由破骨细胞吸收形成初级骨髓腔,成骨细胞在钙化软骨表面形成骨化中心,骨干两端从中央辐射状向四周成骨,长骨两端成为骨骺。骨干与骨骺之间保留一定厚度的软骨层,称为生长板,是长骨增长的基础,至17-20岁时失去增生能力。骨的加长是生长板生长并被骨组织替代的过程,在软骨储备区,软骨细胞较小,为软骨祖细胞(progenitor);软骨增生区,软骨细胞增殖活跃,分裂形成同源细胞群,为前肥大软骨细胞(prehypertrophy);软骨成熟区,软骨细胞变大,为肥大软骨细胞(hypertrophy),软骨基质变薄;软骨钙化区,软骨细胞退化死亡,软骨基质钙化;成骨区钙化的软骨基质表面形成骨组织,同时骨髓腔向两端扩展。成骨细胞在骨干外表面不断添加骨组织,使骨干变粗,破骨细胞在骨干内表面不断吸收骨质是骨髓腔扩大。关于此过程的病理猜测有以下几种:①Keith认为是由于干骺端骨膜的发育不完全,不能完全约束骨骺软骨细胞,致使骨骼组织畸形生长形成骨软骨瘤;②Virchow认为是由于骨骺线的边缘分离出软骨块,以后又长在骨的表面上形成的;③Jansen认为是由于骨骼生长过程中干骺失去塑形能力,使干骺增宽并连续增殖而形成骨疣;④先天性胚胎生长发育缺陷;⑤骨骺板错置移位;⑥由骨膜内层残存幼稚细胞化生而成;⑦酸性黏多糖代谢混乱[2]。 2.4 分子遗传学研究 遗传性多发性骨软骨瘤具有遗传异质性,在分子遗传学的研究中己知遗传性多发性骨软骨瘤与EXT(exotosin)基因突变有关,EXT1 (exotosin-1,MIM # 608997)、EXT2(exotosin-2,MIM #608210)基因分别包含11和16个外显子,于1993和1994年先后被发现并被克隆,位于人类染色体8q23-q24、11p11-p12[11-13],统称为EXT基因。Le Merrer等[14]通过连锁分析,猜测还存在定位于染色体19p的EXT3基因,但EXT3基因至今仍未被克隆。人类遗传性多发性骨软骨瘤患者中有70%-94%发现了EXT1及EXT2突变,可以是单独其中一个基因发生突变或者同时发生突变[15-16],因为EXT1和EXT2基因的突变在遗传性多发性骨软骨瘤患者中具有种系遗传,学者们因此推测它们参与了遗传性多发性骨软骨瘤的发病,而后通过研究mRNA及构建大肠杆菌模型的方式证实EXT基因的肿瘤抑制作用及突变致病性[17-18],随着基因检测技术的进步,原先未能检测出EXT1和EXT2突变的患者被检测出大片段的基因缺失及基因的镶嵌,提出体细胞嵌合体伴有大片基因缺失是EXT基因突变阴性的遗传性多发性骨软骨瘤患者的发病机制,只有极少数患者不能检测出突变,学者们越来越怀疑EXT3的存在[19-21]。以EXT序列作为查询序列在dbEST数据库进行同源性查询,学者们发现了与EXT具有相同羧基末端的高度相似的3个基因,并将其列入EXT基因家族,分别为定位于染色体1p36的EXTL1基因;1p11-p12的EXTL2基因;8p12-p22的EXTL3基因,但并没有EXTL基因导致HME或者其他疾病的证据[22-24]。在欧美国家统计当中,EXT1较EXT2多见,而在中国EXT2突变致病相对更多,说明遗传性多发性骨软骨瘤基因突变具有民族差异[25],在欧美国家报道中,EXT1突变致病病情更为严重。与遗传性多发性骨软骨瘤发病相关的EXT突变位点目前被收录在莱登突变数据库(http://databases.lovd.nl/shared/genes, LOVD),截至2017-09-05,EXT1已有745个突变,EXT2有407个突变,包括碱基置换突变、移码突变、无义突变、缺失突变、插入突变、终止密码突变等多种突变,无热点突变。关于遗传性多发性骨软骨瘤的分子发病机制,学术界有2种假说:①单倍剂量不足:当基因表达水平下降到一个界值时,EXT基因产物单倍剂量不足,导致骨软骨瘤发生;②杂合性缺失:遗传性多发性骨软骨瘤患者遗传了EXT1或者EXT2中任一种功能失调的基因,体细胞中的正常等位基因再次发生突变,最终导致局部骨软骨瘤的产生。 EXT1和EXT2基因具有高度同源性[18],其编码产物广泛存在于所有体细胞当中,分别编码746和718个氨基酸的蛋白质[26],均为定位于高尔基体的Ⅱ型跨膜糖蛋白.二者形成异源寡聚复合物硫酸乙酰肝素多聚酶,从而发挥作用,具有很高的糖基转移酶活性。硫酸乙酰肝素多聚酶作为硫酸乙酰肝素合成酶催化葡萄糖醛酸、N-乙酰葡糖胺交替附着,延伸肽链,形成硫酸乙酰肝素,再与核心蛋白以共价键形式结合,形成硫酸乙酰肝素蛋白多糖[27-28]。在对硫酸乙酰肝素缺失的sog9细胞株的研究中首次发现EXT1突变会导致硫酸乙酰肝素合成异常,而将EXT1导入细胞时,细胞恢复硫酸乙酰肝素合成能力[29]。骨软骨瘤的软骨帽是正常软骨细胞和突变细胞的嵌合体,突变细胞占比1/10,由于突变细胞合成及分泌硫酸乙酰肝素减少,不能形成正常软骨组织中硫酸乙酰肝素的浓度梯度[30-31]。有研究以斑马鱼为实验对象,将突变细胞种植到正常细胞中间时并不会形成骨软骨瘤,种植到边缘部位时则会发展为骨软骨瘤,因此学者们推测正常软骨发育依赖于一定浓度的硫酸乙酰肝素,突变细胞硫酸乙酰肝素分泌能力减弱,当其在正常细胞中央时依赖于正常细胞分泌的硫酸乙酰肝素,可以维持正常发育,但是当其在软骨边缘时,硫酸乙酰肝素浓度无法达到生理需求,就会形成骨软骨瘤[32]。体外间充质细胞培养皿中,加入乙酰肝素酶抑制剂(SST0001),能够强烈抑制软骨形成的现象也说明硫酸乙酰肝素对软骨发育的调节作用[33]。硫酸乙酰肝素是蛋白多糖(基底膜蛋白多糖Perlecan、核心蛋白多糖decorin及共结合蛋白聚糖syndecans)的重要组成成分。蛋白多糖存在于软骨细胞膜表面及细胞外基质,参与信号传导、细胞增殖及分化等过程[29,34-35],硫酸乙酰肝素的改变将不可避免地影响到某些蛋白多糖的功能,其中硫酸乙酰肝素链是基底膜蛋白多糖的主要侧链,硫酸乙酰肝素的合成受限使基底膜蛋白多糖在细胞内的异常定位、大量沉积以及在细胞膜表面、细胞外基质表达减少[36],继而影响其他信号传导等生理过程,扰乱软骨细胞的增殖与分化,导致遗传性多发性骨软骨瘤的发生。遗传性多发性骨软骨瘤的恶性程度与硫酸乙酰肝素水平成负相关,当硫酸乙酰肝素水平下降程度较大时,疾病的外显率趋向于完全,细胞表面相关蛋白聚糖和基质相关蛋白聚糖中硫酸乙酰肝素含量的下降影响了许多对骨骼发育至关重要的信使蛋白质,如印第安刺猬蛋白因子、甲状旁腺激素相关肽、成纤维细胞生长因子、骨形态发生蛋白和Wnt蛋白。这些蛋白质的分布、作用范围、稳定性和对靶细胞的作用发生变化[37-38]。总之,现有研究指向同一个方向:在硫酸乙酰肝素介导的信号传导的某一步骤和/或某一种物质发生了改变,导致遗传性多发性骨软骨瘤发生,但是在硫酸乙酰肝素含量减少之后,骨软骨瘤形成的具体机制却仍然未知。 2.5 蛋白机制及信号通路的研究进展 软骨内骨化是一个极其精密复杂的过程,多种生长因子、细胞因子和信号分子共同调控其发生发展。目前主要有以下几个猜测。 2.5.1 印第安刺猬蛋白因子 印第安刺猬蛋白因子在生长板中调控骨的生长,见图2。该假说以印第安刺猬蛋白因子和甲状旁腺激素相关肽为主要媒介一起形成反馈机制。在生长板中,印第安刺猬蛋白因子由前肥大软骨细胞分泌,在硫酸乙酰肝素蛋白聚糖介导下,印第安刺猬蛋白因子与其受体结合,诱导软骨膜细胞分泌甲状旁腺激素相关肽,甲状旁腺激素相关肽与前肥大软骨细胞表面的受体结合后,延迟前肥大软骨细胞分化,防止其凋亡,该作用程浓度依赖性,随着距产生甲状旁腺激素相关肽的软骨膜细胞的距离增加甲状旁腺激素相关肽的浓度逐渐下降,当下降到一个阈值的时候,周围细胞就会开始肥大化,进而凋亡骨化,以此维持骨轴向生长的特点。当硫酸乙酰肝素蛋白聚糖合成减少时,印第安刺猬蛋白因子不能与其受体结合,甲状旁腺激素相关肽分泌减少,前肥大细胞分化并凋亡,软骨出现异常生长及异位骨化,骨的轴向生长方向改变[39-40]。遗传性多发性骨软骨瘤患者肿瘤切除软骨帽中硫酸乙酰肝素减少伴随分布不均匀,骨软骨瘤细胞中甲状旁腺激素相关肽缺失;在EXT1突变的小鼠胚胎中,印第安刺猬蛋白因子无法与其受体结合,而硫酸乙酰肝素蛋白聚糖的表达可以诱导骨肉瘤细胞的凋亡,充质细胞培养皿中的刺猬蛋白抑制剂HhAntag能以剂量依赖的方式有效阻断软骨形成等现象都支持这一假说[36,41-44]。Koziel等[45]在观察EXT1突变小鼠体细胞时发现:当硫酸乙酰肝素浓度的下降时,印第安刺猬蛋白因子与其受体结合增多,甲状旁腺激素相关肽分泌增多,软骨细胞过度增殖,遗传性多发性骨软骨瘤发生。另一种理论认为印第安刺猬蛋白因子游离体增多并且异常扩散,致使生长板软骨细胞分化紊乱,软骨膜下成骨异常,印第安刺猬蛋白因子扩散区域的改变导致生长板丧失了组织极性,加上骨领的缺陷,导致了病理性生长。软骨细胞进一步增殖并跨过缺陷部位,致使生长异常和骨形成异常,最终导致与正常骨生长方向成角度的骨和软骨的形成[46]。"

2.5.2 成纤维细胞生长因子 成纤维细胞生长因子蛋白家族包含23种蛋白,成纤维细胞生长因子与成纤维细胞生长因子受体的结合均需要特定结构的硫酸乙酰肝素作为共受体参与,以成纤维细胞生长因子2研究较为透彻,硫酸乙酰肝素使成纤维细胞生长因子2与其受体的结合更为牢固,维系成纤维细胞对胶原的附着能力[47-48]。因EXT2突变而致死的小鼠胚胎中,也存在成纤维细胞生长因子无法与其受体结合的现象[49]。而从EXT1突变小鼠体细胞中分离出的成纤维细胞,细胞表面的硫酸乙酰肝素量明显减少,对成纤维细胞生长因子2的反应减弱;在生长因子刺激后,细胞增殖明显少于正常成纤维细胞[47]。成纤维细胞生长因子蛋白直接或者通过与印第安刺猬蛋白因子、骨形态发生蛋白等相互作用间接参与骨骼调控,硫酸乙酰肝素浓度下降后成纤维细胞生长因子功能紊乱能够干扰软骨的分化[50]。 2.5.3 骨形态发生蛋白 骨形态发生蛋白是转化生长因子β超家族的一个亚家族,调节骨骼发育的许多阶段,在软骨形成中可以促进间叶细胞聚集及分化[51]。在Prx1(Peroxiredoxin 1)敲除EXT1的小鼠模型中,间充质凝聚分化延迟并且受损,分化分布区域扩散,间充质中骨形态发生蛋白信号传导结构域和骨形态发生蛋白2免疫反应性的分布同样变宽和扩散;在细胞培养中,突变软骨细胞的软骨形成分化延迟,对外源性骨形态发生蛋白的反应性减弱[52]。骨形态发生蛋白信号传导拮抗剂LDN-193189、Palovarotene全身给药能有效抑制条件性EXT1突变小鼠的骨软骨瘤生长[53-54]。硫酸乙酰肝素拮抗剂(surfen)能够加强骨形态发生蛋白信号及其受体的活性和信号传导,引发过度的细胞反应和骨软骨瘤形成[55]。总之,骨形态发生蛋白既可以通过调节促进印第安刺猬蛋白因子,抑制成纤维细胞生长因子促进骨生长,也可以直接促进软骨细胞增殖,抑制肥大细胞凋亡,但骨形态发生蛋白在遗传性多发性骨软骨瘤中的具体机制仍然不清楚。 2.5.4 Wnt蛋白 在骨骼发育早期,Wnt蛋白诱导β-连环蛋白在细胞质中蓄积,β-连环蛋白易位至细胞核并与转录因子结合,控制下游基因转录以确定间充质祖细胞分化为骨细胞或软骨细胞。Wnt/β-连环蛋白信号调节是软骨细胞和成骨细胞分化以及膜内和软骨内成骨的重要调节通路[56-57]。Wnt蛋白在细胞表面与硫酸乙酰肝素蛋白多糖紧密结合,用于维持疏水性Wnt的溶解度并稳定其活性[58]。Wnt/β-连环蛋白信号通路在维持软骨正常生理活动以及生长板功能中也起重要作用,相对于正常软骨细胞,在遗传性多发性骨软骨瘤患者的骨软骨瘤收集的软骨组织中的细胞,β-连环蛋白很少甚至于没有,骨软骨瘤中的软骨细胞的寿命因此而延长,成为肿瘤细胞[59]。 综上所述,控制软骨细胞增殖和分化的信号分子包括印第安刺猬蛋白因子、骨形态发生蛋白、成纤维细胞生长因子、Wnt蛋白等,EXT基因突变之后,软骨组织中硫酸乙酰肝素水平降低、浓度梯度水平破坏,众多调节通路受损紊乱,本应该轴向生长排列的软骨细胞与生长板成角度生长,最终致病,但是EXT基因的确切作用还未明确,仍有许多问题有待解决,如:①所有组织中均存在硫酸乙酰肝素浓度下降[60],只有骨骼系统表现出临床症状;②所有生长板都存在硫酸乙酰肝素浓度下降,但是不同部位临床表现的解剖学负荷明显不同;③同一个家系中相同基因突变导致的不同患者的临床表现的严重程度明显不同[61],导致这些差异存在的原因还有待研究。 2.6 动物模型 综合现有实验结果,细胞与细胞、细胞与信号分子间的相互作用有赖于机体的整体环境,这与组织和器官类型、发育阶段、细胞种类、其他调节系统及局部因素息息相关[62]。因此在细胞学层面的实验很多时候就不足以满足要求,动物实验因物种差异也存在一些限制,在此总结动物实验所获成果及实验中遇到的问题以供参考。 在20世纪初,学者们通过对果蝇ttv的研究,初步对EXT1基因有了一定的了解,而通过果蝇EXT2同系物sotv的研究,学者们发现ttv或者sotv破坏了果蝇相当于人类刺猬蛋白、Wnt和骨形态发生蛋白信号通路的信号传递[63],且骨形态发生蛋白在胚胎早期起作用,中晚期则由硫酸乙酰肝素蛋白聚糖起主要作用[64]。由于斑马鱼和人类基因有着87%的高度同源性,因而斑马鱼是实验研究很好的对象,在遗传性多发性骨软骨瘤研究中,通过将肿瘤细胞移植在不同部位的实验得出了硫酸乙酰肝素浓度影响肿瘤生长的结论[32]。通过研究非洲爪蟾胚胎的发育过程,学者们发现硫酸乙酰肝素蛋白聚糖可以影响XtSulf1,而XtSulf1在胚胎发生过程中有限制骨形态发生蛋白和成纤维细胞生长因子信号传导的作用[65]。EXT1或者EXT2杂合突变的小鼠不能很好的模拟人类表型,只有部分EXT2杂合突变的小鼠在肋骨上有孤立的骨软骨瘤样肿瘤,纯合突变的小鼠由于在胚胎时期不能形成原肠胚而夭折[41,50]。通过Cre重组酶可以条件性阻断EXT1基因复制,使小鼠出现包含少量EXT1纯合突变的EXT1杂合突变,成功构建了骨软骨瘤症状明显的EXT1杂合突变的小鼠模型,为小鼠实验创造了可能,该实验还证实了肿瘤起源于生长板软骨细胞而非Ranvier骨化槽[66-67]。之后又有别的实验通过别的阻断机制构建了小鼠模型,但是绝大多数都是阻断了EXT1的功能性复制,少有EXT2的阻断。这些实验都有一个共同的指向:不论是通过杂合性缺失、二次打击还是通过其他途径,硫酸乙酰肝素含量的减少都是疾病发生的必要条件[52,68]。"

| [1]Solomon L. Hereditary multiple exostosis. Am J Hum Genet. 1964;16: 351-363.[2]Jones KB. Glycobiology and the growth plate: current concepts in multiple hereditary exostoses. J Pediatr Orthop. 2011;31(5): 577-586.[3]Tahasildar N, Sudesh P, Goni V, et al. Giant osteochondroma of axis in a child with multiple hereditary exostoses: case report and review of literature. J Pediatr Orthop B. 2012;21(3): 280-285.[4]Franch J, Font J, Ramis A, et al. Multiple cartilaginous exostosis in a Golden Retriever cross-bred puppy. Clinical, radiographic and backscattered scanning microscopy findings. Vet Comp Orthop Traumatol. 2005;18(3):189-193.[5]Kyriazoglou AI, Dimitriadis E, Arnogiannaki N, et al. Similar cytogenetic findings in two synchronous secondary peripheral chondrosarcomas in a patient with multiple osteochondromas. Cancer Genet. 2011;204(12): 677-681.[6]郭永成,王永堂,陆亚东,等. 儿童遗传性多发性骨软骨瘤软骨帽的超微结构观察[J]. 临床与实验病理学杂志, 2013, 29(2): 171-174.[7]Huvos AG, Marcove RC. Chondrosarcoma in the young. A clinicopathologic analysis of 79 patients younger than 21 years of age. Am J Surg Pathol. 1987; 11(12): 930-942.[8]Legeai-Mallet L, Munnich A, Maroteaux P, et al. Incomplete penetrance and expressivity skewing in hereditary multiple exostoses. Clin Genet. 1997;52(1):12-16.[9]肖福英,蒋林彬,刘健翔,等. 遗传性多发性骨软骨瘤一家系11例[J]. 中华医学遗传学杂志,2005,22(5):574.[10]Rozeman LB, de Bruijn IH, Bacchini P, et al. Dedifferentiated peripheral chondrosarcomas: regulation of EXT-downstream molecules and differentiation-related genes. Mod Pathol. 2009; 22(11): 1489-1498.[11]Cook A, Raskind W, Blanton SH, et al. Genetic heterogeneity in families with hereditary multiple exostoses. Am J Hum Genet. 1993;53(1):71-79.[12]Wu YQ, Heutink P, de Vries BB, et al. Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11. Hum Mol Genet. 1994;3(1): 167-171.[13]Wuyts W, Ramlakhan S, Van Hul W, et al. Refinement of the multiple exostoses locus (EXT2) to a 3-cM interval on chromosome 11. Am J Hum Genet. 1995; 57(2): 382-387.[14]Le Merrer M, Legeai-Mallet L, Jeannin PM, et al. A gene for hereditary multiple exostoses maps to chromosome 19p. Hum Mol Genet. 1994;3(5): 717-722.[15]Jennes I, Pedrini E, Zuntini M, et al. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat. 2009;30(12): 1620-1627.[16]Ishimaru D, Gotoh M, Takayama S, et al. Large-scale mutational analysis in the EXT1 and EXT2 genes for Japanese patients with multiple osteochondromas. BMC Genet.2016;17:52.[17]Ahn J, Lüdecke HJ, Lindow S, et al. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat Genet. 1995;11(2): 137-143.[18]Wuyts W, Van Hul W, Wauters J, et al. Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet. 1996;5(10): 1547-1557.[19]Szuhai K, Jennes I, de Jong D, et al. Tiling resolution array-CGH shows that somatic mosaic deletion of the EXT gene is causative in EXT gene mutation negative multiple osteochondromas patients. Hum Mutat. 2011; 32(2): E2036-2049.[20]Li Y, Wang J, Tang J, et al. Heterogeneous spectrum of EXT gene mutations in Chinese patients with hereditary multiple osteochondromas. Medicine (Baltimore). 2018;97:e12855.[21]Scl S, Impo R, Takata RI, et al. Analysis of mutations in EXT1 and EXT2 in Brazilian patients with multiple osteochondromas. Mol Genet Genomic Med. 2018;6:382-392.[22]Wise CA, Clines GA, Massa H, et al. Identification and localization of the gene for EXTL, a third member of the multiple exostoses gene family. Genome Res. 1997;7(1): 10-16.[23]Wuyts W, Van Hul W, Hendrickx J, et al. Identification and characterization of a novel member of the EXT gene family, EXTL2. Eur J Hum Genet. 1997;5(6): 382-389.[24]Van Hul W, Wuyts W, Hendrickx J, et al. Identification of a third EXT-like gene (EXTL3) belonging to the EXT gene family. Genomics. 1998;47(2): 230-237.[25]Kang QL, Xu J, Zhang Z, et al. Mutation screening for the EXT1 and EXT2 genes in Chinese patients with multiple osteochondromas. Arch Med Res. 2013;44(7): 542-548.[26]Stancheva-Ivanova MK, Wuyts W, van Hul E, et al. Clinical and molecular studies of EXT1/EXT2 in Bulgaria. J Inherit Metab Dis. 2011;34(4): 917-921.[27]Lindahl U. Heparan sulfate-protein interactions--a concept for drug design. Thromb Haemost. 2007;98(1): 109-115.[28]Li JP. Heparin, heparan sulfate and heparanase in cancer: remedy for metastasis. Anticancer Agents Med Chem. 2008. 8(1): 64-76.[29]McCormick C, Leduc Y, Martindale D, et al. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet. 1998;19(2): 158-161.[30]de Andrea CE, Prins FA, Wiweger MI, et al. Growth plate regulation and osteochondroma formation: insights from tracing proteoglycans in zebrafish models and human cartilage. J Pathol. 2011; 224(2): 160-168.[31]Hameetman L, David G, Yavas A, et al. Decreased EXT expression and intracellular accumulation of heparan sulphate proteoglycan in osteochondromas and peripheral chondrosarcomas. J Pathol. 2007;211(4): 399-409.[32]Clément A, Wiweger M, der Hardt SV, et al. Regulation of zebrafish skeletogenesis by ext2/dackel and papst1/pinscher. PLoS Genet. 2008;4(7): e1000136.[33]Huegel J, Enomoto-Iwamoto M, Sgariglia F, et al. Heparanase stimulates chondrogenesis and is up-regulated in human ectopic cartilage: a mechanism possibly involved in hereditary multiple exostoses. Am J Pathol. 2015;185:1676-1685.[34]Schwartz NB, Domowicz M. Chondrodysplasias due to proteoglycan defects. Glycobiology. 2002;12(4): 57R-68R.[35]Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001; 11(2): 75-82.[36]Hecht JT, Hall CR, Snuggs M, et al. Heparan sulfate abnormalities in exostosis growth plates. Bone. 2002; 31(1): 199-204.[37]Zak BM, Schuksz M, Koyama E, et al. Compound heterozygous loss of Ext1 and Ext2 is sufficient for formation of multiple exostoses in mouse ribs and long bones. Bone. 2011;48(5): 979-987.[38]Jones KB, Pacifici M, Hilton MJ. Multiple hereditary exostoses (MHE): elucidating the pathogenesis of a rare skeletal disorder through interdisciplinary research. Connect Tissue Res. 2014;55(2): 80-88.[39]Duncan G, McCormick C, Tufaro F. The link between heparan sulfate and hereditary bone disease: finding a function for the EXT family of putative tumor suppressor proteins. J Clin Invest. 2001; 108(4): 511-516.[40]Beltrami G, Ristori G, Scoccianti G, et al. Hereditary Multiple Exostoses: a review of clinical appearance and metabolic pattern. Clin Cases Miner Bone Metab. 2016;13(2): 110-118.[41]Lin X, Wei G, Shi Z, et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol. 2000;224(2): 299-311.[42]Hameetman L, Kok P, Eilers PH, et al. The use of Bcl-2 and PTHLH immunohistochemistry in the diagnosis of peripheral chondrosarcoma in a clinicopathological setting. Virchows Arch. 2005;446(4): 430-437.[43]Modrowski D, Orosco A, Thévenard J, Fromigué O, Marie PJ. Syndecan-2 overexpression induces osteosarcoma cell apoptosis: Implication of syndecan-2 cytoplasmic domain and JNK signaling. Bone. 2005;37(2): 180-189.[44]Mundy C, Bello A, Sgariglia F, et al. HhAntag, a Hedgehog Signaling Antagonist, Suppresses Chondrogenesis and Modulates Canonical and Non-Canonical BMP Signaling. J Cell Physiol. 2016;231:1033-1044.[45]Koziel L, Kunath M, Kelly OG, et al. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell. 2004; 6(6): 801-813.[46]Benoist-Lasselin C, de Margerie E, Gibbs L, et al. Defective chondrocyte proliferation and differentiation in osteochondromas of MHE patients. Bone. 2006;39(1): 17-26.[47]Osterholm C, Barczyk MM, Busse M, et al. Mutation in the heparan sulfate biosynthesis enzyme EXT1 influences growth factor signaling and fibroblast interactions with the extracellular matrix. J Biol Chem. 2009; 284(50): 34935-34943.[48]Häcker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005; 6(7): 530-541.[49]Shimokawa K, Kimura-Yoshida C, Nagai N, et al. Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell. 2011;21(2): 257-272.[50]Stickens D, Zak BM, Rougier N, et al. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 2005; 132(22): 5055-5068.[51]Huegel J, Mundy C, Sgariglia F, et al. Perichondrium phenotype and border function are regulated by Ext1 and heparan sulfate in developing long bones: a mechanism likely deranged in Hereditary Multiple Exostoses. Dev Biol. 2013; 377(1): 100-12.[52]Matsumoto Y, Matsumoto K, Irie F, et al. Conditional ablation of the heparan sulfate-synthesizing enzyme Ext1 leads to dysregulation of bone morphogenic protein signaling and severe skeletal defects. J Biol Chem. 2010; 285(25): 19227-19234.[53]Sinha S, Mundy C, Bechtold T, et al. Unsuspected osteochondroma-like outgrowths in the cranial base of Hereditary Multiple Exostoses patients and modeling and treatment with a BMP antagonist in mice. PLoS Genet. 2017; 13(4): e1006742.[54]Inubushi T, Lemire I, Irie F, et al. Palovarotene Inhibits Osteochondroma Formation in a Mouse Model of Multiple Hereditary Exostoses. J Bone Miner Res. 2018;33:658-666.[55]Mundy C, Yang E, Takano H, et al. Heparan sulfate antagonism alters bone morphogenetic protein signaling and receptor dynamics, suggesting a mechanism in hereditary multiple exostoses. J Biol Chem, 2018;293:7703-7716.[56]Day TF, Guo X, Garrett-Beal L, et al. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5): 739-750.[57]Sgariglia F, Pedrini E, Bradfield JP, et al. The type 2 diabetes associated rs7903146 T allele within TCF7L2 is significantly under-represented in Hereditary Multiple Exostoses: insights into pathogenesis. Bone. 2015;72:123-127.[58]Kikuchi A, Yamamoto H, Sato A, et al. New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011; 291: 21-71.[59]Yuasa T, Kondo N, Yasuhara R, et al. Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 2009;175(5): 1993-2003.[60]Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007; 446(7139): 1030-1037.[61]Pedrini E, Jennes I, Tremosini M, et al. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: identification of "protective" and "risk" factors. J Bone Joint Surg Am. 2011;93(24): 2294-2302.[62]Rider CC, Mulloy B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem J. 2010;429(1): 1-12.[63]Bornemann DJ, Duncan JE, Staatz W, et al. Abrogation of heparan sulfate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development. 2004;131(9): 1927-1938.[64]Bornemann DJ, Park S, Phin S, et al. A translational block to HSPG synthesis permits BMP signaling in the early Drosophila embryo. Development. 2008;135(6): 1039-1047.[65]Freeman SD, Moore WM, Guiral EC, et al. Extracellular regulation of developmental cell signaling by XtSulf1. Dev Biol. 2008; 320(2): 436-445.[66]Jones KB, Piombo V, Searby C, et al. A mouse model of osteochondromagenesis from clonal inactivation of Ext1 in chondrocytes. Proc Natl Acad Sci U S A. 2010;107(5): 2054-2059.[67]Nozawa S, Inubushi T, Irie F, et al. Osteoblastic heparan sulfate regulates osteoprotegerin function and bone mass. JCI Insight. 2018; 3(3). pii: 89624.[68]Matsumoto K, Irie F, Mackem S, et al. A mouse model of chondrocyte-specific somatic mutation reveals a role for Ext1 loss of heterozygosity in multiple hereditary exostoses. Proc Natl Acad Sci U S A. 2010; 107(24): 10932-10937. |
| [1] |
Cai Miaoxin, Yang Bin, Deng Peng, Ge Yingjie, Guo Fuming, Liang Zhenghui, Zhai Pei, Pang Zhihui, Fan Yueguang.
A three-dimensional femoral head necrosis classification system based on biplanar (frontal and frog-leg lateral) X-ray images: Biomechanics Laboratory of Guangzhou University of Chinese Medicine Classification system
[J]. Chinese Journal of Tissue Engineering Research, 2019, 23(24): 3786-3791.
|
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||